Ewing-Sarkom – Kurzinformation
Das Ewing-Sarkom ist ein bösartiger Tumor des Kindes- und Jugendalters. In diesem Text erhalten Sie die wichtigsten Informationen zu Krankheitsbild, Häufigkeit, möglichen Krankheitsverläufen, Ursachen und Symptomen, zu Diagnose, Therapieplanung und Behandlung sowie zur Prognose der Erkrankung.
Autor: Maria Yiallouros, Dr. med. habil. Gesche Tallen, Redaktion: Maria Yiallouros, Freigabe: Prof. Dr. med. Uta Dirksen, Prof. Dr. md. U. Creutzig, Zuletzt geändert: 16.04.2024 https://dx.doi.org/10.1591/poh.patinfo.ewing.kurz.20101215
Inhaltsverzeichnis
Krankheitsbild
Ewing-Sarkome sind solide bösartige Tumoren, die meist im Knochen auftreten. Selten entstehen sie in Weichteilgeweben, also in Binde-, Fett-, Muskelgewebe oder Gewebe peripherer Nerven. Die Erkrankung ist nach dem New Yorker Krebsforscher James Ewing (1866-1943) benannt, der diesen Tumor im Jahre 1921 beschrieb. Die meisten Ewing-Sarkome wachsen und streuen sehr schnell, so dass die Erkrankung ohne eine wirksame Behandlung tödlich verläuft.
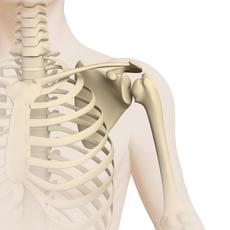
Jeder Knochen kann Ursprungsort eines Ewing-Sarkoms sein. Am häufigsten betroffen ist das Becken, gefolgt von den langen Röhrenknochen der Ober- und Unterschenkel, den Rippen, dem Schulterblatt und der Wirbelsäule.
Die Tumoren können sich sowohl innerhalb des Knochens als auch in den umgebenden Weichgeweben ausbreiten. In seltenen Fällen (circa 15 %) entstehen Ewing-Sarkome direkt in den Weichteilen, also außerhalb und ohne Beteiligung des Knochens. Man spricht in diesem Fall von extraskelettalen oder extraossären Ewing-Tumoren. Reine Weichteil-Ewing-Sarkome können beispielsweise in den Nieren, den Nebennieren, der Lunge oder im Magen-Darm-Trakt vorkommen.
Ewing-Sarkome wachsen schnell und bilden frühzeitig Tochterabsiedlungen (Metastasen). Bei etwa einem Viertel der Patienten liegen bereits zum Zeitpunkt der Diagnosestellung sichtbare Metastasen vor, meist in der Lunge, aber auch in Knochen und Knochenmark. Darüber hinaus haben fast alle Patienten kleinste Metastasen – so genannte Mikrometastasen – die mit herkömmlichen Untersuchungsverfahren noch nicht zu erkennen sind. Ewing-Sarkome gelten daher als Erkrankungen, die den ganzen Körper betreffen (Systemerkrankung).
Häufigkeit
Ewing-Sarkome sind (nach den Osteosarkomen) die zweithäufigsten Knochentumoren bei Kindern und Jugendlichen. Sie machen insgesamt etwa 2 % aller Krebserkrankungen im Kindes- und Jugendalter aus. In Deutschland erkranken nach Angaben des Deutschen Kinderkrebsregisters (Mainz) jährlich etwa 50 Kinder und Jugendliche unter 18 Jahren (das entspricht drei pro Million) neu an einem Ewing-Sarkom.
Das Ewing-Sarkom kann in jedem Alter auftreten, seinen Häufigkeitsschwerpunkt hat es jedoch im zweiten Lebensjahrzehnt. In der Altersgruppe der 0-17-Jähringen erkranken besonders häufig Jugendliche zwischen 12 und 17 Jahren. Die Erkrankung kommt aber auch im Säuglings-, Kleinkind- und Schulkindalter vor. Das durchschnittliche Erkrankungsalter (Altersgruppe 0-17) liegt bei etwa 13 Jahren. Jungen und männliche Heranwachsende sind häufiger betroffen als Mädchen (Geschlechterverhältnis 1,3:1).
Feingewebliche Eigenschaften und Tumortypen
Ewing-Sarkome gehören zu den primitiven bösartigen Tumoren. Es ist bis heute noch nicht bekannt, aus welcher Ursprungszelle sie hervorgehen. Nach dem derzeitigen Stand der Forschung entwickeln sie sich aus unreifen (undifferenzierten) Gewebezellen, so genannten mesenchymalen Stammzellen, oder aus primitiven, neuroektodermalen Stammzellen.
Ewing-Sarkomzellen werden feingeweblich (histologisch) den mesenchymalen klein-, blau- und rundzelligen Tumoren zugeordnet; sie können nur durch spezielle immunhistochemische und molekulargenetische Untersuchungen von undifferenzierten Tumorzellen anderer Krebserkrankungen (wie Neuroblastom, Medulloblastom, Non-Hodgkin-Lymphom, Weichteilsarkomen, kleinzelligem Osteosarkom und Retinoblastom) unterschieden werden. Wegen der Seltenheit der Tumoren erfolgen die entsprechenden Untersuchungen in darauf spezialisierten Laboren (siehe Abschnitt "Diagnose").
Bis vor einiger Zeit wurden – anhand von feingeweblichen Eigenschaften und dem Ursprungsort des Tumors – innerhalb der Gruppe der Ewing-Sarkome verschiedene Tumortypen unterschieden. Dazu gehörten das klassische Ewing-Sarkom (EWS), der periphere maligne primitive neuroektodermale Tumor (PPNET oder pPNET), der Askin-Tumor der Brustwand und der Ewing-Tumor der Weichteile. Nach der aktuellen Einteilung der Weltgesundheitsorganisation (WHO-Klassifikation) werden die verschiedenen Tumoren zu einer Entität zusammengefasst, das heißt, es ist generell nur noch der Begriff „Ewing-Sarkom“ vorgesehen. Alle Ewing-Sarkome sind hochgradig bösartig.
Ursachen
Die Ursachen für die Entstehung eines Ewing-Sarkoms sind unbekannt. Weder äußere Einflussfaktoren, wie zum Beispiel eine vorangegangene Strahlentherapie, noch vererbte genetische Faktoren (erbliche Veranlagung) scheinen eine wesentliche Rolle zu spielen. Allerdings zeigt die Erkrankung eine ethnische Präferenz, das heißt, sie tritt bei Angehörigen der hellhäutigen (kaukasischen) Bevölkerung deutlich häufiger auf als bei Asiaten und Afrikanern.
Bekannt ist auch, dass die Tumorzellen der Ewing-Sarkome gewisse Chromosomenveränderungen aufweisen, die immer ein bestimmtes Gen auf Chromosom 22 – das so genannte Ewing-Sarkom-Gen (EWS-Gen) – einschließen. Die Veränderungen entstehen durch einen Austausch von Chromosomenabschnitten (Translokation), meist zwischen dem EWS-Gen auf Chromosom 22 und einem Gen auf Chromosom 11. Die mit 85 % häufigste Translokation [genannt t(11;22) (q24;q12)-Translokation] ist so typische für Ewing-Sarkome, dass ihr Nachweis die Diagnose der Erkrankung ermöglicht. In vielen Ewing-Sarkomen sind darüber hinaus weitere genetische Veränderungen bekannt. Die aus solchen Abweichungen resultierenden Gendefekte sind daran beteiligt, dass aus einer gesunden Zelle eine Tumorzelle wird. Generell werden solche im Tumorgewebe nachweisbaren Genveränderungen aber nicht vererbt.
Sehr selten tritt das Ewing-Sarkom Zusammenhang mit einem Krebsprädispositionssyndrom, einer erblich bedingten Veranlagung für die Entwicklung von Tumoren, oder als Zweitkrebserkrankung (Sekundärmalignom) auf. Letzteres kann auch noch viele Jahre nach Abschluss der Behandlung einer ersten (primären) Krebserkrankung vorkommen.
Krankheitszeichen
Die bei weitem häufigsten Beschwerden, die durch ein Ewing-Sarkom verursacht werden, sind Schmerzen und eine Schwellung in der vom Tumor betroffenen Region.
Die Schmerzen können unregelmäßig auftreten und sind gewöhnlich aktivitätsbezogen, gehen aber oft auch während der Nacht nicht ganz zurück. Zu den Schmerzen kann – mit zunehmendem Tumorwachstum – eine sichtbare und/oder tastbare, eventuell gerötete Schwellung hinzukommen, die mit Funktionseinbußen einhergehen kann. Nicht selten werden diese Krankheitszeichen zunächst als Wachstumsschmerzen, eine Knochenentzündung oder als Folge einer Sportverletzung fehlgedeutet.
Da Ewing-Sarkome praktisch in jedem Knochen und in Weichgewebe entstehen können, sind die weiteren Symptome von Patient zu Patient verschieden. Sind zum Beispiel die Wirbelsäule und/oder periphere Nerven [peripheres Nervensystem] betroffen, können Ausfallerscheinungen wie Lähmungen im Vordergrund stehen. Tumoren der Becken- oder Brustregion oder auch Tumoren im Oberschenkel können lange Zeit unbemerkt bleiben. Bei etwa einem Drittel der Patienten treten Allgemeinsymptome wie Fieber, Krankheitsgefühl, Gewichtsverlust und/oder eine allgemeine Müdigkeit auf, die auf eine bereits fortgeschrittene Erkrankung hinweisen können. Von den ersten Symptomen bis zur endgültigen Diagnose der Erkrankung können einige Wochen oder Monate vergehen.
Gut zu wissen: Kinder und Jugendliche mit Beschwerden, wie sie hier beschrieben sind, haben selbstverständlich nicht immer ein Ewing-Sarkom oder einen anderen bösartigen Knochentumor. Dennoch ist es ratsam, jede Form von Knochenschmerzen im Kindes- und Jugendalter sorgfältig durch einen erfahrenen Kinderarzt abklären zu lassen, um eine bösartige Erkrankung auszuschließen.
Diagnose
Findet der (Kinder-)Arzt durch Krankheitsgeschichte (Anamnese) und körperliche Untersuchung Hinweise auf einen bösartigen Knochentumor, wird er den Patienten in ein Krankenhaus überweisen, das auf Krebserkrankungen bei Kindern und Jugendlichen spezialisiert ist (Klinik für pädiatrische Onkologie/Hämatologie). Denn bei Verdacht auf einen solchen Tumor sind umfangreiche Untersuchungen und die Zusammenarbeit von Spezialisten unterschiedlicher Fachrichtungen notwendig, um festzustellen, ob tatsächlich ein bösartiger Knochentumor vorliegt und, wenn ja, um welche Form des Tumors es sich handelt und wie weit sich die Erkrankung im Körper ausgebreitet hat. Die Klärung dieser Fragen ist Voraussetzung für eine optimale Behandlung und Prognose des Patienten.
Laboruntersuchungen
Zur Diagnose eines Ewing-Sarkoms gehören, neben einer erneuten Anamnese-Erhebung und körperlichen Untersuchung, die Untersuchung von Blut und Urin. Es gibt zwar keine Tumormarker, die spezifisch ein Ewing-Sarkom anzeigen könnten, aber bestimmte Auffälligkeiten, die im Rahmen dieser Laboruntersuchungen festgestellt werden, können erste Hinweise auf die Art der Erkrankung geben und/oder die Abgrenzung zu anderen in Frage kommenden Tumorerkrankungen unterstützen.
Bildgebende Untersuchungen zum Tumornachweis
Der Verdacht auf einen bösartigen Knochentumor kann meist bereits anhand typischer Befunde im Röntgenbild [Röntgenuntersuchung] erhärtet werden. Mit Hilfe zusätzlicher bildgebender Verfahren wie der Magnetresonanztomographie (MRT) und/oder der Computertomographie (CT) mit Kontrastmittel lassen sich die genaue Lage und Größe des Tumors sowie seine Abgrenzung zu Nachbarstrukturen (wie Muskel- und Sehnengewebe oder Gelenkkapseln) sehr gut darstellen. Auch nahe gelegene Metastasen – so genannte Skip-Metastasen – sind mit diesen Methoden gut sichtbar.
Die MRT ist bei der Bestimmung betroffener Weichteil- und Knochenmarkanteile der CT überlegen, so dass dieses Verfahren neben der Röntgenübersichtsaufnahme des betroffenen Knochens bevorzugt bei der Erstdiagnose des Tumors eingesetzt wird. Sie dient auch als Grundlage für die spätere Planung der Operation und für die Überwachung des Krankheitsverlaufes während der Chemotherapie. Eine CT kann allerdings in seltenen Fällen zusätzlich erforderlich sein, um Veränderungen im Knochen genauer zu untersuchen.
Entnahme und Untersuchung von Tumorgewebe
Um die Diagnose eines Ewing-Sarkoms endgültig zu sichern, muss aber in jedem Fall eine Gewebeprobe entnommen werden. Die Gewebeentnahme (Biopsie) sollte immer von Ärzten durchgeführt werden, die auch auf die Operation von Sarkomen spezialisiert sind. Damit wird sichergestellt, dass der für die Biopsie gewählte Zugang später nicht zu Problemen bei der weiteren Behandlung führt. Eine ungünstig geplante Biopsie kann dazu führen, dass eine spätere Operation sehr viel größer ausfallen muss als eigentlich notwendig wäre oder, schlimmstenfalls, ein eigentlich operabler Tumor nach einer ungeeigneten Operation nicht mehr operabel ist.
Die Gewebeentnahme erfolgt entweder im Rahmen einer Operation am freigelegten Tumor (offene Biopsie) oder als Stanzbiopsie von außen. Im letzteren Fall werden mit Hilfe spezieller Nadeln mehrere Gewebezylinder aus dem Tumor gestanzt. Die entnommenen Gewebeproben werden anschließend von mehreren Spezialisten sowohl feingeweblich (histologisch) als auch immunhistochemisch und molekulargenetisch untersucht. Die molekulargenetische Untersuchung ist von besonderer Bedeutung, weil der Nachweis einer für Ewing-Sarkome typischen genetischen Veränderung (siehe Abschnitt „Ursachen“) das Vorliegen eines Ewing-Sarkoms bestätigt und die Abgrenzung von anderen, ähnlichen Tumorarten ermöglicht.
Untersuchungen zur Ausbreitung der Erkrankung (Stadieneinteilung)
Wenn die Diagnose „Ewing-Sarkom“ feststeht, erfolgen weitere Untersuchungen zur Klärung der Krankheitsausbreitung. Auch hier spielen bildgebende Verfahren eine maßgebende Rolle. Erforderlich ist zunächst eine genaue Vermessung des Primärtumors (so genannte Volumetrie), da dessen Volumen (und im Laufe der Behandlung dessen Verkleinerung) für die Heilungsaussichten des Patienten von Bedeutung ist.
Zum Nachweis oder Ausschluss von Lungenmetastasen wird eine Computertomographie des Brustkorbs (CT-Thorax) durchgeführt. Eine Skelett-Szintigraphie mit schwach radioaktiv markiertem Technetium (Tc99) dient der Suche nach Knochenmetastasen. Zunehmend wird anstelle der Skelett-Szintigraphie auch die sehr sensitive Positronen-Emissions-Tomographie (PET) mit 18-Fluor-Deoxyglukose, einem radioaktiv markierten Zucker, eingesetzt (FDG-PET). Die nuklearmedizinische Methode wird in diesem Fall immer mit einer CT oder einer MRT kombiniert (CT-PET / MRT-PET). Unabhängig davon, ob eine Skelett-Szintigraphie oder eine PET gewählt wird, erfolgt zudem eine MRT aller klinisch und nuklearmedizinisch verdächtiger Regionen. In Einzelfälle kann auch eine Ganzkörper-MRT nützlich sein.
Um festzustellen, ob das Knochenmark befallen ist, müssen auch eine Knochenmarkpunktion sowie Knochenmarkstanzen an mehreren Stellen erfolgen (Knochenmarkstanzbiopsie). Das entnommene Knochenmark wird feingeweblich und molekulargenetisch untersucht. Bei Verdacht auf einen Befall des Zentralnervensystems ist möglicherweise auch eine Lumbalpunktion erforderlich. Je nach nach Krankheits- und Behandlungssituation können weitere Untersuchungen hinzukommen.
Behandlungsvorbereitende Untersuchungen
Vor Beginn der Behandlung erfolgen eine Überprüfung der Herzfunktion (Elektrokardiographie [EKG] und Echokardiographie), der Hörfunktion (Audiometrie) und der Nieren- und Lungenfunktion sowie verschiedene Blutuntersuchungen. Veränderungen, die möglicherweise im Laufe der Therapie auftreten, können aufgrund solcher Ausgangsbefunde besser beurteilt und bei der Behandlung entsprechend berücksichtigt werden.
Gut zu wissen: Nicht alle Untersuchungen sind bei jedem Patienten notwendig. Andererseits können eventuell Untersuchungen hinzukommen, die hier nicht erwähnt wurden. Fragen Sie Ihren behandelnden Arzt oder das Behandlungsteam, welche Untersuchungen bei Ihrem Kind geplant sind und warum die jeweilige Untersuchung erforderlich ist.
Therapieplanung
Nachdem die Diagnose feststeht, erfolgt die Therapieplanung. Um eine möglichst individuelle, auf den Patienten zugeschnittene (risikoadaptierte) Behandlung durchführen zu können, berücksichtigt das Behandlungsteam bestimmte Faktoren, die die Prognose des Patienten beeinflussen (so genannte Risiko- oder Prognosefaktoren).
Wichtige Prognosefaktoren bei Patienten mit einem Ewing-Sarkom sind zum einen die Art, Lage, Größe und Ausdehnung des Tumors (lokal begrenzt oder metastasiert), die anhand der beschriebenen Diagnoseverfahren ermittelt werden. Darüber hinaus sind aber auch das Ausmaß der operativen Tumor-/Metastasenentfernung (unvollständig oder vollständig) und das Ansprechen der Erkrankung auf die Chemotherapie von entscheidender Bedeutung. Alle diese Faktoren fließen in die Behandlungsplanung ein mit dem Ziel, für jeden Patienten das jeweils bestmögliche Behandlungsergebnis zu erreichen.
Therapie
Die Behandlung eines Patienten mit Ewing-Sarkom muss in einer kinderonkologischen Behandlungseinrichtung erfolgen. Dort ist das hoch qualifizierte Fachpersonal (Ärzte, Fachpflegekräfte) auf die Behandlung krebskranker Kinder spezialisiert und mit den modernsten Therapieverfahren vertraut. Die Ärzte dieser Klinikabteilungen stehen in fachorientierten Arbeitsgruppen in ständiger, enger Verbindung miteinander und behandeln ihre Patienten nach gemeinsam entwickelten und stetig weiter verbesserten Therapieplänen. Optimal ist die Behandlung in einer Einrichtung mit einem ausgewiesenen Sarkomschwerpunkt, ansonsten sollte die Kinderonkologie mit einer solchen zusammenarbeiten, vor allem auch für Operation und Strahlentherapie.
Das Ziel der Behandlung ist, eine Heilung des Patienten zu erreichen und dabei das Risiko therapiebegleitender Nebenwirkungen und Spätfolgen so gering wie möglich zu halten.
Behandlungsmethoden
Bei Patienten mit einem Ewing-Sarkom besteht die Behandlung prinzipiell aus einer Lokaltherapie (Operation und/oder Strahlentherapie) und einer Chemotherapie. Bei manchen Patienten kann, unter bestimmten Voraussetzungen, auch eine Hochdosis-Chemotherapie mit anschließender autologer Stammzelltransplantation in Frage kommen.
Die Strahlentherapie erfolgt durch den Einsatz energiereicher Strahlen (Photonen oder Protonen), die von außen durch die Haut auf die betroffene Region eingestrahlt werden. Sie verursachen Schäden im Erbgut der Tumorzellen und führen dadurch zu deren Absterben. Bei der Chemotherapie werden zellwachstumshemmende Medikamente (Zytostatika) verabreicht, die darauf abzielen, Krebszellen in ihrem Wachstum zu stoppen oder zu vernichten.
Mit Operation und Bestrahlung wird die maximal mögliche lokale Kontrolle der Erkrankung angestrebt. Die zusätzliche Chemotherapie ist wichtig, weil sich gezeigt hat, dass allein mit einer Operation und/oder Bestrahlung der Tumor zwar oft entfernt werden kann, später jedoch fast immer Metastasen auftreten. Daher ist eine Behandlung erforderlich, die – wie die Chemotherapie – den ganzen Körper betrifft (so genannte systemische Therapie). Allerdings kann die Chemotherapie allein die Lokaltherapie nicht ersetzen. Für manche Patienten mit hohem Rückfallrisiko kann die Hochdosis-Chemotherapie und die anschließende autologe Stammzelltransplantation eine zusätzliche Option darstellen.
Da die Behandlung eines Ewing-Sarkoms mit akuten Nebenwirkungen einhergehen kann, erfolgen während der Behandlung unterstützende Therapiemaßnahmen (Supportivtherapie), die der Vorbeugung und/oder Behandlung dieser Begleiteffekte dienen. Hier finden Sie Informationen zur Supportivtherapie sowie Empfehlungen für zu Hause.
Behandlungsablauf
In der Regel erfolgt die Lokaltherapie zwischen zwei Chemotherapiephasen. Die gesamte Behandlung dauert etwa zehn Monate, kann aber auch – abhängig von vielen Faktoren – mehr Zeit erfordern. Folgende Therapiephasen werden unterschieden:
Chemotherapiephase vor der Lokaltherapie
Die Behandlung beginnt bei allen Patienten mit einer mehrwöchigen intensiven Chemotherapie (auch Induktions-Chemotherapie oder Induktionstherapie genannt). Das Ziel dieser Chemotherapie ist, den Tumor und eventuell vorhandene Metastasen zu verkleinern und abzutöten und auf diese Weise die nachfolgende Operation schonender und sicherer und damit so effektiv wie möglich zu machen. Darüber hinaus dient die Chemotherapie der Bekämpfung kleinster, noch nicht sichtbarer Tochterabsiedlungen (Mikrometastasen) und soll verhindern, dass der Tumor weiter streut.
Um möglichst alle bösartigen Tumorzellen zu vernichten, wird eine Kombination verschiedener zellwachstumshemmender Medikamente (Zytostatika) eingesetzt, die sich bei der Bekämpfung von Ewing-Sarkomen als besonders wirkungsvoll erwiesen haben. Hierzu gehören zum Beispiel die Medikamente Vincristin, Doxorubicin (= Adriamycin), Cyclophosphamid, Ifosfamid und Etoposid (VDC/IE). Die Zytostatika werden in mehreren (aktuell neun) mehrtägigen Chemotherapiezyklen als Infusion verabreicht. In dieser Zeit wird der Patient in die Klinik aufgenommen. In den dazwischenliegenden Therapiepausen kann der Patient in der Regel zu Hause sein; eine erneute stationäre Aufnahme ist nur bei schweren Nebenwirkungen erforderlich.
Lokaltherapie – Operation und Bestrahlung
Noch während oder spätestens im Anschluss an die Chemotherapie erfolgt die Lokaltherapie. Die bevorzugte Behandlung ist die Operation mit dem Ziel, den Tumor möglichst vollständig zu entfernen. (Dieser chirurgische Eingriff sollte unbedingt in einem spezialisierten Sarkomzentrum erfolgen.) Eine komplette Tumorentfernung ist allerdings aufgrund der Lage des Tumors in funktionell wichtigen Körperregionen manchmal nicht möglich, so dass ergänzend oder stattdessen eine Strahlentherapie durchgeführt wird. Eine Kombination aus Operation und Strahlentherapie ist in einigen Fällen notwendig, da die kombinierte Behandlung das Risiko für einen Krankheitsrückfall senkt.
Welche der beiden Behandlungsmethoden in Frage kommt oder ob beide Verfahren kombiniert werden, hängt vom einzelnen Patienten und seiner Erkrankungssituation ab und muss ganz individuell entschieden werden. Das Behandlungsteam wird Sie über Art und Ablauf des chirurgischen Eingriffs beziehungsweise über die Strahlentherapie genauer informieren. Bei Tumoren der Arme oder Beine ist es dank der großen Fortschritte im Bereich der Gliedmaßen erhaltenden Operationstechniken und durch Einsatz von Chemo- und/oder Strahlentherapie heute oft möglich, auf eine Amputation zu verzichten.
Im Anschluss an eine Operation untersucht der Pathologe das entnommene Ewing-Sarkom um festzustellen, wie gut die Erkrankung auf die vorangegangene Chemotherapie angesprochen hat. Dies wird am Anteil der noch verbliebenen lebenden Tumorzellen gemessen. Liegt der Anteil unter 10 %, so spricht man von einem guten Tumoransprechen; beträgt der Anteil der noch lebenden Tumorzellen 10 % oder mehr, spricht man von einem schlechten Tumoransprechen. Das Tumoransprechen (definiert nach den Graden 1-6) wird bei der weiteren Behandlung mitberücksichtigt.
Gut zu wissen: Zum Zeitpunkt der Diagnose vorhandene Metastasen werden, wenn möglich, wie der Primärtumor lokal behandelt, also operativ entfernt und/oder bestrahlt.
Chemotherapiephase nach der Lokaltherapie
Nach der Lokaltherapie wird die Chemotherapie fortgesetzt (sie wird dann auch als Konsolidierungs-Chemotherapie oder Konsolidierungstherapie bezeichnet). Die Intensität der Behandlung richtet sich einerseits nach der Größe und Ausdehnung des Tumors zum Zeitpunkt der Diagnose und andererseits danach, wie gut der Tumor auf die bereits vor der Operation durchgeführte Chemotherapie angesprochen hat.
Patienten mit lokalisierter Erkrankung und schlechtem Ansprechen auf die Induktions-Chemotherapie (mehr als 10 % lebende Tumorzellen) oder mit einem großen Tumor (Volumen ab 200 ml) erhalten nach der Konsolidierungstherapie, wenn möglich, eine Hochdosis-Chemotherapie (mit Busulfan und Melphalan) und und im Anschluss eine autologe Stammzelltransplantation. Diese Behandlung führt, wie sich gezeigt hat, zu besseren Behandlungsergebnissen, setzt aber voraus, dass die Konsolidierung zu einer vollständigen Tumorrückbildung (Remission) geführt hat.
Liegen zum Zeitpunkt der Diagnose bereits Lungenmetastasen vor und bilden sich diese durch die Konsolidierungstherapie vollständig zurück (komplette Remission), schließt sich eine zusätzliche Bestrahlung der gesamten Lunge an. Eine Hochdosis-Chemotherapie mit anschließender autologer Stammzelltransplantation zeigt bei diesen Patienten keinen Vorteil gegenüber der Standard-Chemotherapie. Letzteres gilt in der Regel auch für Patienten, die zum Diagnosezeitpunkt (noch) andere Metastasen als Lungenmetasen aufweisen. Sie erhalten daher meist eine konventionelle Chemotherapie; lediglich bei einer Untergruppe von Kindern bis zum 14. Lebensjahr kann die Hochdosis-Chemotherapie und autologe Stammzelltransplantation von Vorteil sein.
Behandlung bei Krankheitsrückfall
Trotz verbesserter Therapiemethoden erleiden noch immer 30 bis 40 % der Patienten mit Ewing-Sarkom einen Krankheitsrückfall (Rezidiv). Eine Standardtherapie-Empfehlung gibt es für diese Patienten derzeit nicht. Je nach Krankheitssituation können eine Chemotherapie mit mehreren Medikamenten (zum Beispiel Topoisomerasehemmer wie Etoposid, Irinotecan oder Topotecan und Alkylantien wie Ifosfamid, Cyclophosphamid und Temozolomid), eine Strahlentherapie, chirurgische Maßnahmen oder eine Kombination dieser Methoden erwogen werden. Auch eine Hochdosis-Chemotherapie kann bei Erreichen einer kompletten Remission nach Rezidivtherapie in Frage kommen.
Wenn eine Behandlung mit dem Ziel der Heilung nicht mehr möglich ist, steht die Erhaltung der Lebensqualität des Patienten im Vordergrund, zum Beispiel durch Schmerzlinderung und den Erhalt von Funktionen (Palliativtherapie). Im Rahmen von Phase-I-/II-Studien wird versucht, die Heilungsaussichten auch dieser Patienten zu verbessern, zum Beispiel durch Einsatz und Erprobung neuer Medikamente.
Therapieoptimierungsstudien und Register
In den großen Behandlungszentren der Welt werden Kinder und Jugendliche mit einem Ewing-Sarkom nach standardisierten Behandlungsprotokollen behandelt. Sie alle haben zum Ziel, die Langzeitüberlebensraten der Patienten zu verbessern und gleichzeitig therapiebedingte Spätfolgen so gering wie möglich zu halten. Die Behandlung nach solchen Therapieprotokollen erfolgt in aller Regel im Rahmen von Therapieoptimierungsstudien oder Registern.
Therapieoptimierungsstudien sind kontrollierte klinische Studien, die darauf abzielen, erkrankte Patienten nach dem jeweils aktuellsten Wissensstand zu behandeln und gleichzeitig die Behandlungsmöglichkeiten zu verbessern und weiter zu entwickeln. Patienten, die an keiner Studie teilnehmen, entweder weil zum Zeitpunkt ihrer Erkrankung keine Studie verfügbar ist oder weil sie die Einschlusskriterien einer bestehenden Studie nicht erfüllen, werden oft in einem so genannten Register dokumentiert. Diese dienen zunächst dazu, die Therapie der Patienten wissenschaftlich zu begleiten. Zur Sicherung der optimalen Behandlung verfasst darüber hinaus die jeweilige Studiengruppe in der Regel detaillierte Empfehlungen und berät die behandelnden Ärzte bei der Auswahl der optimalen Therapie für den einzelnen Patienten.
In Deutschland können seit 2020 Kinder und Jugendliche (sowie Erwachsene) mit erstmaliger Erkrankung an einem Ewing-Sarkom, die nicht im Rahmen von Studien behandelt werden können, in das internationale Euro Ewing Register aufgenommen werden. Das Register stellt eine Fortführung des Mitte 2019 geschlossenen EWING 2008-Registers dar. Die Studienkoordination für Deutschland befindet sich an der Klinik für Kinder- und Jugendheilkunde der Universität Essen (Studienleitung: Prof. Dr. med. Uta Dirksen).
Für Patienten mit Rückfall eines Ewings-Sarkoms besteht in Deutschland seit Ende 2018 die Möglichkeit, an der internationalen Phase-II-Studie rEECur teilzunehmen. Im Rahmen dieser randomisierten, kontrollierten Studie werden vier verschiedene Zytostatikakombinationen (Topotecan und Cyclophosphamid; Irinotecan und Temozolomid; Gemcitabine und Docetaxel; Hochdosis-Ifosfamid) miteinander verglichen. Dabei soll festgestellt werden, welcher Chemotherapie-Ansatz zu den besten Therapieergebnissen führt. Leiter der Studie ist Dr. Martin McCabe (Birmingham). Die Studienkoordination für Deutschland befindet sich an der Klinik für Kinder- und Jugendheilkunde der Universität Essen (Studienleitung: Prof. Dr. med. Uta Dirksen).
Patienten mit Krankheitsrückfall können sich darüber hinaus in das INFORM-Register aufnehmen lassen. Das Register erfasst systematisch Erbgutveränderungen in den Tumoren mit dem Ziel, für Rückfallpatienten zukünftig eine individuell zugeschnittene Behandlung anbieten und damit die Heilungschancen verbessern zu können. INFORM steht für INdividualized Therapy FOr Relapsed Malignancies in Childhood oder (deutsch) Individualisierte Therapie für Rückfälle von bösartigen Tumoren bei Kindern. Die Studienkoordination befindet sich am Hopp-Kindertumorzentrum Heidelberg (KiTZ) unter der Leitung von Prof. Dr. med. Olaf Witt.
Prognose
Die Prognose von Kindern und Jugendlichen mit einem Ewing-Sarkom hängt von verschiedenen Faktoren ab. Entscheidend sind insbesondere die Lage und Größe des Tumors, seine Ausbreitung zum Zeitpunkt der Diagnosestellung und das Ansprechen der Erkrankung auf die präoperative Chemotherapie.
In den letzten Jahrzehnten haben sich dank der Einführung intensiver Kombinationschemotherapien und der standardisierten Behandlung im Rahmen von Therapieoptimierungsstudien die Überlebensaussichten der Patienten mit Ewing-Sarkom deutlich verbessert.
Während die Überlebenschance in den 1960er-Jahren mit alleiniger Strahlentherapie oder Operation bei weniger als 10 % lag, können heute durch die Kombination von lokaler Therapie und Chemotherapie durchschnittlich über 80 % der Patienten mit lokalisierter Erkrankung, das heißt, ohne sichtbare Metastasen, langfristig von dieser Erkrankung geheilt werden. Voraussetzung für eine günstige Prognose ist in der Regel, dass der Tumor vollständig entfernt werden kann und die Erkrankung gut auf die Chemotherapie anspricht.
Patienten, deren Erkrankung zum Zeitpunkt der Diagnose bereits metastasiert ist, haben trotz intensiver chemotherapeutischer Behandlung nach wie vor eine ungünstige Prognose (5-Jahres-Überlebensraten von durchschnittlich etwa 20 bis 25 %). Dabei sind die Überlebensaussichten für Patienten mit einzelnen, operablen Lungenmetastasen besser als die für Patienten mit Knochen- oder Knochenmarkmetastasen. Ähnlich ungünstige Heilungsaussichten haben Patienten, die einen Krankheitsrückfall (Rezidiv) erleiden. Am ungünstigsten ist die Prognose für Patienten, die nach einer intensiven Erstbehandlung frühzeitig Metastasen entwickeln. Im Rahmen aktueller und zukünftiger Studien wird versucht, die Prognose auch für diese Patienten zu verbessern.
Anmerkung: Bei den genannten Überlebensraten handelt es sich um statistische Größen. Sie stellen nur für die Gesamtheit der an einem Ewing-Sarkom erkrankten Patienten eine wichtige und zutreffende Aussage dar. Ob der einzelne Patient geheilt werden kann oder nicht, lässt sich aus der Statistik nicht vorhersagen. Der Begriff Heilung muss hier vor allem als „Tumorfreiheit“ verstanden werden. Denn auch wenn die heute verfügbaren Therapiemethoden zu langfristiger Tumorfreiheit führen können, so sind sie doch häufig auch mit unerwünschten Nebenwirkungen und Spätschäden verbunden, die in der Regel eine intensive Rehabilitation und eine langfristige orthopädische Betreuung erforderlich machen.
 PDF-Datei der Patienten-Kurzinformation zum Ewing-Sarkom (456KB)
PDF-Datei der Patienten-Kurzinformation zum Ewing-Sarkom (456KB)
Autor: Maria Yiallouros, Dr. med. habil. Gesche Tallen
Stand: 09.11.2022
Literaturliste 
- Zöllner SK, Amatruda JF, Bauer S, Collaud S, de Álava E, DuBois SG, Hardes J, Hartmann W, Kovar H, Metzler M, Shulman DS, Streitbürger A, Timmermann B, Toretsky JA, Uhlenbruch Y, Vieth V, Grünewald TGP, Dirksen U: Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives. Journal of clinical medicine 2021 Apr 14; 10 [PMID: 33919988]
- Erdmann F, Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Annual Report 2019 (1980-2018). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2020 [URI: https://www.kinderkrebsregister.de/ typo3temp/ secure_downloads/ 42507/ 0/ 1c5976c2ab8af5b6b388149df7182582a4cd6a39/ Buch_DKKR_Jahresbericht_2019_komplett.pdf]
- Bauer S, Dirksen U, Schildhaus HU: [Systemic therapy of sarcomas : New biomarkers and therapeutic strategies]. Der Pathologe 2019, 40: 436 [PMID: 31243550]
- Dirksen U, Brennan B, Le Deley MC, Cozic N, van den Berg H, Bhadri V, Brichard B, Claude L, Craft A, Amler S, Gaspar N, Gelderblom H, Goldsby R, Gorlick R, Grier HE, Guinbretiere JM, Hauser P, Hjorth L, Janeway K, Jürgens H, Judson I, Krailo M, Kruseova J, Kuehne T, Ladenstein R, Lervat C, Lessnick SL, Lewis I, Linassier C, Marec-Berard P, Marina N, Morland B, Pacquement H, Paulussen M, Randall RL, Ranft A, Le Teuff G, Wheatley K, Whelan J, Womer R, Oberlin O, Hawkins DS, Euro-EWING 99 and Ewing 2008 Investigators: High-Dose Chemotherapy Compared With Standard Chemotherapy and Lung Radiation in Ewing Sarcoma With Pulmonary Metastases: Results of the European Ewing Tumour Working Initiative of National Groups, 99 Trial and EWING 2008. Journal of clinical oncology 2019,JCO1900915 [PMID: 31553693]
- Whelan J, Le Deley MC, Dirksen U, Le Teuff G, Brennan B, Gaspar N, Hawkins DS, Amler S, Bauer S, Bielack S, Blay JY, Burdach S, Castex MP, Dilloo D, Eggert A, Gelderblom H, Gentet JC, Hartmann W, Hassenpflug WA, Hjorth L, Jimenez M, Klingebiel T, Kontny U, Kruseova J, Ladenstein R, Laurence V, Lervat C, Marec-Berard P, Marreaud S, Michon J, Morland B, Paulussen M, Ranft A, Reichardt P, van den Berg H, Wheatley K, Judson I, Lewis I, Craft A, Jürgens H, Oberlin O, Euro-EWING99 and EWING-2008 Investigators: High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E. W.I. N.G. 99 and Ewing-2008. J Clin Oncol 2018 [PMID: 30188789]
- Scobioala S, Ranft A, Wolters H, Jabar S, Paulussen M, Timmermann B, Jürgens H, Hassenpflug W, Klingebiel T, Elsayad K, Eich HT, Dirksen U: Impact of Whole Lung Irradiation on Survival Outcome in Patients With Lung Relapsed Ewing Sarcoma. International journal of radiation oncology, biology, physics 2018 Nov 1; 102: 584 [PMID: 30244879]
- Whelan J, Hackshaw A, McTiernan A, Grimer R, Spooner D, Bate J, Ranft A, Paulussen M, Jürgens H, Craft A, Lewis I: Survival is influenced by approaches to local treatment of Ewing sarcoma within an international randomised controlled trial: analysis of EICESS-92. Clinical sarcoma research 2018, 8: 6 [PMID: 29610659]
- Dirksen U, Jürgens H: Ewing-Sarkom. in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie, Springer-Verlag GmbH Deutschland, 2. vollständig überarbeitete Auflage 2018, 497 [ISBN: 978-3-662-43685-1]
- Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, Kovar H, Grimer R, Whelan J, Claude L, Delattre O, Paulussen M, Picci P, Sundby Hall K, van den Berg H, Ladenstein R, Michon J, Hjorth L, Judson I, Luksch R, Bernstein ML, Marec-Bérard P, Brennan B, Craft AW, Womer RB, Jürgens H, Oberlin O: Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015 Sep 20; 33: 3036 [PMID: 26304893]
- Haeusler J, Ranft A, Boelling T, Gosheger G, Braun-Munzinger G, Vieth V, Burdach S, van den Berg H, Jürgens H, Dirksen U: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 2010, 116: 443 [PMID: 19924786]
- Burkhardt B, Reiter A, Landmann E, Lang P, Lassay L, Dickerhoff R, Lakomek M, Henze G, von Stackelberg A: Poor outcome for children and adolescents with progressive disease or relapse of lymphoblastic lymphoma: a report from the berlin-frankfurt-muenster group. Journal of clinical oncology 2009, 27: 3363 [PMID: 19433688]
- Gerth HU, Juergens KU, Dirksen U, Gerss J, Schober O, Franzius C: Significant benefit of multimodal imaging: PET/CT compared with PET alone in staging and follow-up of patients with Ewing tumors. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2007, 48: 1932 [PMID: 18006618]
- Wagner LM, McAllister N, Goldsby RE, Rausen AR, McNall-Knapp RY, McCarville MB, Albritton K: Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatric blood & cancer 2007, 48: 132 [PMID: 16317751]
- Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jürgens H: Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatric blood & cancer 2006, 47: 795 [PMID: 16411206]
- Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA, Jürgens H: Ewing's sarcoma family of tumors: current management. The oncologist 2006, 11: 503 [PMID: 16720851]
- Hawkins DS, Schuetze SM, Butrynski JE, Rajendran JG, Vernon CB, Conrad EU 3rd, Eary JF: [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005, 23: 8828 [PMID: 16314643]
- Dirksen U, Poremba C, Schuck A: Knochentumoren des Kindes-und Jugendalters. Der Onkologe 2005, 10: 1034
- Schuck A, Ahrens S, Paulussen M, Kuhlen M, Konemann S, Rube C, Winkelmann W, Kotz R, Dunst J, Willich N, Jürgens H: Local therapy in localized Ewing tumors: results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int J Radiat Oncol Biol Phys 2003, 55: 168 [PMID: 12504050]