Nöroblastom (Kısa Bilgiler)
Nöroblastom sıklıkla erken çocukluk çağında görülen ve sempatik sinir sisteminden kaynaklanan kötü huylu bir tümördür. Bu yazıda hastalığın ortaya çıkışı, hastalığın seyri, sıklığı, hastalığa sebep olan nedenler, semptomları, tanısı, tedavi planlaması, tedavisi ve hastalığın prognozu hakkında bilgiler bulunmaktadır.
yazar: Maria Yiallouros, editör: Maria Yiallouros, Yayın İzni: Prof. Dr. med. Frank Berthold, Prof. Dr. med. Angelika Eggert, Prof. Dr. med. Thorsten Simon, türk tercüman: Dr. med. Ebru Saribeyoglu, Sait Kont, Last modification: 2024/04/16 https://dx.doi.org/10.1591/poh.neurobl.patinfo.kurz.1.20120611
Table of contents
Hastalık tablosu
Nöroblastomlar kötü huylu solid tümörlerdir. Bunlar sempatik sinir sisteminin kontrolden çıkmış ilkel hücrelerinden ortaya çıkarlar. Sempatik sinir sistemi, otonom sinir sistemin bir parçası olarak kalp atışı, kan dolaşımı, bağırsak ve mesanenin (idrar torbası) işlevi gibi vücudun istem dışı fonksiyonlarının yönlendirilmesinden sorumludur.
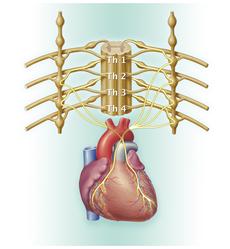
Nöroblastomlar sempatik sinir dokularının bulunduğu her yerde ortaya çıkabilirler. En sık görüldükleri yerler böbreküstü bezi dokusu ve omurganın her iki tarafındaki sinir ağlarındaki trunkus sempatikus isimli alanlardır. Trunkus sempatikusun tutulduğu durumlarda, nöroblastomlar omurganın her iki tarafında ve herhangi bir bölgesinde belirebilirler. Örneğin karın, kalça, göğüs ve boyunda nöroblastomlar oluşabilir. Vakaların çoğunda (yaklaşık %70 kadarında) tümörler karın bölgesinde belirmektedir, buna karşın tümörlerin yaklaşık beşte biri göğüs ve boyun bölgelerinde görülmektedir.
Hastaların yaklaşık yarısında nöroblastom oluştuğu yerde sınırlı kalmaktadır, diğer yarısı ise kendisine yakın lenf düğümlerine (lenf bezlerine, lenf bezelerine) sıçramaktadır. Bazı hastalarda kemik iliğinde, kemikte, uzaktaki lenf düğümlerinde, karaciğerde veya ciltte, nadiren de beyinde veya akciğerde hastalıklı hücreler yani metastazlar belirebilmektedir. Nöroblastomların kendilerine has bir özelliği de, spontan olarak kendiliklerinden küçülüp ortadan kaybolabilmeleridir.
Görülme sıklığı
Çocuk ve gençlerde görülen tüm kanserlerin yaklaşık % 5,5 kadarını nöroblastomlar oluşturmaktadır. Nöroblastomlar, merkezi sinir sistemi (MSS tümörleri, beyin tümörleri) tümörlerinden sonra bu yaş grubunda en sık rastlanan solid tümörlerdir. Mainz Alman çocuk kanserleri veri bankasının açıklamalarına göre Almanya’da her sene 18 yaş altındaki yaklaşık 120 tane çocuk ve genç nöroblastom vakasına rastlanmaktadır. Dolayısıyla her sene 18 yaş altı 1.000.000 çocuktan yaklaşık birinde bu hastalık görülmektedir.
Nöroblastomlar embriyonal yani olgunlaşmamış hücrelerden oluşan tümörler olduklarından, bunlara özellikle küçük çocuk yaşlarında rastlanır: Hastaların % 90 kadarı 6 yaşından küçüktür. Hastaların çoğunluğu (yaklaşık % 46’sı) yeni doğanlar ve süt çocukluğu evresindeki bebeklerdir. Erkek çocuklarda kızlara oranla biraz daha sıkça görülmektedir (erkek kız oranı: 1,4/1). Öte yandan nöroblastomlar büyük çocuklarda ve geçlerde ve hatta nadiren yetişkinlerde de belirebilmektedir.
Sebepleri
Nöroblastomun oluşma sebebi henüz tam net olarak bilinmemektedir. Bilinen şu dur ki nöroblastom hastalığı sempatik sinir sistemindeki ilkel hücrelerin bazılarının dejenerasyona (değişime) uğrayarak habisleşmesi sebebiyle oluşur. Bu henüz olgunlaşmamış (embriyonal) sinir hücrelerinin hatalı gelişmesi, muhtemelen henüz bebek doğmadan başlamaktadır. Bunun sebepleri kromozom değişiklikleri ve/veya hatalı gen regülasyonu olabilir.
Nöroblastom tümör hücrelerinde bir çok genetik değişiklikler saptanmıştır, ancak bu değişiklikler çok heterojendir, yani tüm tümörlerde görülebilen spesifik kalıtsal değişiklikler saptanamamıştır. Sonuç olarak nöroblastom gelişiminde bir çok genetik (kalıtsal) ve epigenetik değişiklikler rol oynamaktadır. Güncel bilimsel veriler ışığında günümüzde hastaların çoğunluğunda hastalığın kalıtsal olmadığı anlaşılmaktadır.
Ancak nöroblastomların ve benzer tümörlerin bir çok jenerasyonda görüldüğü aileler vardır. Tüm neuroblastom hastaların %1-2 sinde bu durum söz konusudur ve sıklıkla birden fazla birincil tümörleri vardır. Bunun dışında nöroblastom, kansere yatkınlık yaratan sendromları (kalıtsal olarak tümör oluşma sıklığının artmış olması) ile ilişkili olabilmektedir. Nöroblastom gelişimi ile ilgili olan kanser yatkınlık sendromları örneğin Morbus Hirschsprung veya Undine sendromu olabilir.
Hastaların büyük çoğunluğunda hastalık spontan mutasyonlar veya vücut hücrelerindeki diğer genomik değişikiliklerin bir sonucu olarak ortaya çıkar. Dış etkenlerin, (örneğin çevresel faktörlerin, ebeveynlerin mesleki yüklerinin, ilaç kullanımının, hamilelik döneminde sigara veya alkol kullanımının) hastalığın oluşmasında bir rol oynayıp oynamadığı konusu bugüne kadar netlik kazanmamıştır.
Hastalık belirtileri
Nöroblastomlu hastaların hastalığa özgü hastalık belirtileri (semptomlari) yoktur. Tümöre çoğunlukla örneğin çocuk doktorunda bir rutin muayenede veya diğer sebeplerle yapılan bir ultrasonografi veya röntgen tetkiki sırasında şans eseri rastlanmaktadır. Şikayetler çoğu zaman ancak tümör ileri derecede büyüdükten, kardeş tümör (metastaz) oluşturduktan veya çevredeki organ ve dokulara olumsuz etkiler başladıktan sonra görülür.
Nöroblastom hastalık belirtileri çok çeşitlidir. Bunlar tümörün veya metastazların bulunduğu yerlere bağlı olarak değişmektedir. Elle dokunarak farkedilebilen tümörler ve metastazlar hastalığın ilk belirtileri olabilir. Karın boşluğu veya böbreküstü bezi kaynaklı tümörlerde karın ağrısı, doygunluk/şişlik hissi veya ishal ortaya çıkabilir. Tümörün idrar yollarına basısı sonucu idrar akışında bozulma (idrar birikmesi) görülebilir. Eğer tumor göğüs kafesi içinde ise yaptığı bası sonucu öksürük, akciğer iltihabı veya solunum sıkıntısı bulguları oluşabilir. Omurilik çevresinde büyüyen tümörler, omurilik kanalı içine yayılım göstererek nörolojik bulgulara (örneğin sinir ağrısı, idrar ve dışkı çıkışında sorunlar veya hatta felç bulgularına) yol açabilirler.
Bazı ender vakalarda tümörün hormon aktivitesine bağlı olarak tansiyon yükselebilir veya kronik ishal oluşabilir. Boyun bölgesindeki tümörler, Horner sendromu denilen duruma neden olabilir. Bu sendromda, göz küresinin tek taraflı küçülmüş göz bebeğiyle ve sarkık göz kapağıyla birlikte içeriye çökmesi halidir. Bunun yanısıra göz bölgesinde örneğin göz kapağı ekimozu denilen (göz kapağı çevresinde morluklar) diğer değişiklikler olabilir. Hastalığın ilerlemiş devrelerinde gözlerin etrafında bazen halka şeklinde hematomlar oluşur (monokel hematomu, Rakun gözü). Hastalığın ender bir seyir şekli de opsomiyoklonus ataksi sendromudur (OMAS).
Özellikle kol ve bacak gibi uzun kemiklerdeki veya göz çevresi ve kafatasındaki metastazlar kemik ağrılarına sebep olabilirler. Bazı hastalarda ağrılar kendini sadece hafif bir topallama ile gösterebilir. Eğer kemik iliğinde yaygın bir tutulum da söz konusu ise kansızlık (anemi), kan pulcuklarında azalma (trombositopeni) ve beyaz kan hücreleinde azalma (lökopeni) ve bunlara bağlı infeksiyonlar veya kanamaya eğilimler ortaya çıkabilir.
Nöroblastom hastalığını akla getirecek (sıklıkla da ilerlemiş olabileceğini gösteren) genel semptomlar şunlardır:
- Yorgunluk, isteksizlik, güçsüzlük, solukluk ve performans azalması
- Belirgin bir sebebi olmaksızın sürekli hafif ateş, terleme
- Karın veya boyunda bezeler veya şişkinlikler; lenf bezlerinde şişkinlikler
- Şişkin büyük karın
- Kabızlık ve ishal, karın ağrısı
- İştahsızlık, bulantı, kusma; bunlara bağlı kilo kaybı
- Kemik ağrıları
- Gözlük hematomu
Bilinmesi gereken nokta: Yukarıda sayılan hastalık belirtilerinin birinin veya birkaçının görülmesi mutlaka bir nöroblastom söz konusu olduğu anlamına gelmez. Bu semptomların bir çoğu, nöroblastoma kıyasla önemsiz nedenlerden kaynaklanabilir. Ama öte yandan herhangi bir şikayet ve rahatsızlık durumunda, en kısa zamanda nedenlerini anlamak için bir doktora danışılması tavsiye edilir.
Tanı
Doktor veya çocuk doktoru, çocuğun hastalık geçmişinde (anamnezi) ve/veya bedensel muayene yani fiziksel muayenesi sırasında bir nöroblastom olabileceğine dair veriler elde ederse, hastayı çocuk ve gençlerde kanser ve kan hastalıkları uzmanı bir hastaneye sevk edecektir (Pediatrik Onkoloji / Hematoloji Kliniği). Çünkü böyle bir tümör şüphesi durumunda teşhisi kesinleştirmek ve hangi hastalık türüne sahip olduğu konusunda bir sonuca varmak için çeşitli tanısal işlemler gereklidir. Ancak bu araştırmalar yapıldıktan sonra uygun bir tedavi ve prognoz (tedavi başarısı) mümkün olabilir.
Laboratuvar testleri
Tanının konulmasında önce laboratuvar tetkikleri önemli bir rol oynar. Nöroblastomlu hastaların çoğunun kanında veya idrarında tümör belirteçleri (tümör işaretleyicisi) diye tanımlanan vücuda özgü bazı maddelere yüksek değerlerde rastlanır. Bunlar hem tanı konulmasında hem de özellikle hastalığın seyri süresince uygulanan tedavinin başarısını kontrol etmede kullanılabilecek verilerdir. Nöroblastomlarda önemli sayılan tümör markerlerinden bazıları katekolaminler veya bunların artık maddeleri (dopamin, vanilya asidi, homo vanilik asit ) ve nöron spesifik enolaz [nöron spesifik enolaz, NSE] maddesidir.
Görüntüleme yöntemleri
Tanının kanıtlanmasına ve nöroblastomun örneğin Wilms tümörü veya feokromasitoma gibi diğer hastalıklarından ayırt edilmesi amacıyla görüntüleme yöntemlerinden faydalanılır: ultrasonografi yyardımıyla çoğu nöroblastomun yerleşim yeri ve büyüklüğü, hatta boyun/batın ve pelvis içinde lenf bezi tutulumu olup olmadığı saptanabilir. Röntgen [röntgen] ekilerek akciğerler ve göğüs kafesi değerlendirilebilir.
Çok ufak tümörleri de bulabilmek ve bunların etraflarındaki oluşumlara (örneğin diğer organlara, kan damarlarına, sinirlere) yayılıp yayılmadıklarını anlamak için ayrıca bir manyetik rezonans tomografisi (MR) uygulanır. Bazı seçilmiş hastalarda MR yerine bilgisayarli tomografi (BT) daha uygun olabilir.
Metastaz tarama tetkikleri
Metastazların kanıtlanması veya dışlanabilmesi için ve primer tümörün daha iyi değerlendirilebilmesi için hafif radyoaktif madde içeren 123iyot-meta-iyotbenzilguanin kullanılarak, kısaca IMBG sintigrafi çekilebilir. MIBG sintigrafisi ile sonuç alınamazsa alternatif olarak radyoaktif işaretli şekerli madde ile (18 flor deoksiglukoz, kısaca FDG) pozitron emisyon tomografisi (kısaca PET). Her iki yöntem bilgisayarlı tomografi (BT) veya manyetik rezonans görüntüleme (MR) ile kombine edilir.
Hastalık kemik iliğine henüz çok az yerleşmişse, bunun sintigrafi yoluyla tespit edilmesi mümkün olmadığından, her hastadan dört değişik bölgeden kemik iliği örneği alınıp incelenmesi gerekmektedir. Kemik iliği örneği, kemik iliği ponksiyonu veya kemik iliğinin zımbalı biyopsisi yoluyla alınır. Bu girişim genellikle kısa süreli anestezi altında gerçekleştirilir. Alınan örnek mikroskop altında ve özel metodlarla kötü huylu hücreler bulunup bulunmadığı yönünde incelenir. Metastaz bulunan hastalarda, hastalığın beyine sıçrayıp sıçramadığını saptayabilmek için kafatasının MRT muayenesi yapılır. Ayrıca böyle ilerlemiş vakalarda tüm vücut MR incelemesi yapılarak kemik metastazlarının da aranması gerekebilir.
Doku örneğin alınıp incelenmesi (biyopsi)
Tanının kanıtlanması prensip olarak ancak tümörlü dokudan örnek alıp bunun patolojik mikroskop altında (histolojik) incelenmesi ile mümkün olabilir. Tümörlü dokudan örnek alınması genellikle cerrahi girişim (operasyon) yoluyla gerçekleşir. Alınan dokuda yapılacak moleküler genetik incelemeler tümörün kötü huyluluk derecesi (habislik) ile ilgili bilgi verir. Tümördeki DNA içinde saptanan bazı değişiklikler (mutasyon), örneğin MYCN amplifikasyonu veya 1p delesyonu, hastalık seyri (prognozu) konusunda olumsuz gidiş anlamına gelebilir. Öte yandan burada sözü edilen değişikliklerin görülmemesi veya diğer mutasyonlar, olumlu prognoz anlamına gelebilmektedir. İşte bu konularda kesin bir sonuca varabilmek için, moleküler genetik incelemelerin yapılması büyük önem taşımaktadır.
Yakın zaman önce nöroblastom hücreleri üzerinde bazı gen defektleri (hataları) tanımlanmıştır (örneğin ALK-geninde değişiklikler veya telomeraz aktivasyonu gibi). Bu gen defektlerinin bulunması örneğin hastalığın tekrar etmesi (nüks etmesi) durumunda tedavi yöntemi olarak da kullanılabilir.
Tedaviye hazırlık incelemeleri
Yapılması planlanan tedavinin türüne göre henüz tedavi başlamadan bazı organların durum ve fonksiyonlarını kontrol edebilmek için diğer ek tetkikler yapılır. Bu tetkikler şunlar olabilir: Kemoterapi öncesinde kalp muayenesi (elektrokardiyografi [EKG], ekokardiyografi), işitme muayenesi (odyometri), böbrek fonksiyon testleri veya böbrek ultrasonografisi. Ayrıca çocuğun elinin röntgen incelemesi çocuğun büyüme durumu hakkında bir görüşe varılır. Elde edilen başlangıç verileri, tedavi süresince ortaya çıkabilecek değişikliklerin daha iyi değerlendirilmesine yardımcı olur ve tedavide dikkate alınır.
Bilinmesi gereken nokta: Yukarıda sayılan tetkiklerin hepsini her hastada uygulamaya gerek olmayabilir. Öte yandan burada sayılmayan diğer başka tetkiklerin de yapılması gerekli olabilir. Çocuğunuz için hangi tetkiklerin planlandığını ve neden gerekli olduklarını doktorunuza veya tedavi ekibinize sorunuz.
Tedavi planı
Tanı kesinleştikten sonra tedavi planlanır. Tedaviyi planlayan ekip mümkün olduğunca kişiye özgü yani hastaya göre uyarlanmış (risk adaptasyonu yapılmış) bir tedavinin gerçekleştirilmesi amacıyla hastadaki prognoz durumunu (tedaviye yanıtı) etkileyen belirli faktörleri (risk ve prognoz faktörleri) dikkate alır.
Bu çerçevede hastanın hangi hastalık evresinde bulunduğu çok önemlidir. Hastalık evresini anlayabilmek için tanı anında tümörün ne ölçüde vücuda yayıldığına ve ameliyat ile tamamen çıkarılıp çıkarılamayacağına bakılır (alttaki evreleme tablosuna bakınız). Diğer önemli prognozu etkileyen faktörler; hastanın yaşı, tümörün mikroskopik ve moleküler genetik özellikleridir („teşhis-tanı“ bölümüne bakınız). Tüm bu faktörler tedavi planı yapılırken göz önünde tutulur. Bunun amacı, hasta için mümkün olan en iyi ve uygun tedavi yöntemini bulmaktır.
Nöroblastomun evreleri hakkında ayrıntılı bilgileri aşağıda bulacaksınız.
Hastalık evreleri
Hastalığın vücuttaki yayılımı iyileşme oranını doğrudan etkilediği için uygulanacak tedavinin seçiminde çok önemli bir kriterdir. Nöroblastomların evrelendirilmesinde tümörün büyüklüğü, lenf bezlerinde tutulum olup olmaması ve tümörün metastaz yapıp yapmadığı göz önüne alınır. Evrelendirmede hangi kriterlerin göz önüne alınması gerektiği kullanılan evrelendirme yöntemine göre farklılık gösterebilir. Halen iki evreleme sistemi parallel olarak kullanılmaktadır: INSS evreleme sistemi ve ınrg evreleme sistemi.
- INSS-Evreleme sistemi: Almanyada uzun yıllardır uluslarası olarak da kabul görmüş (uluslararası nöroblastom evreleme sistemi-INSS) ve yukarıda sayılan faktörlere ek olarak uygulanan cerrahi girişimin boyutunu da kriter olarak kullanan bu sistem kullanılmaktadır. Ancak bu evreleme sisteminde, tam evreleme cerrahi girişim sonrası yapılabilmektedir.
- INRG-Evreleme sistemi: Yukarıda bahsedilen INSS evreleme sistemi gözönünde bulundurulmasına rağmen günümüzde daha çok uluslararası nöroblastom risk grup evreleme sistemi-INRG) kabul görmektedir. INRG evreleme sisteminde cerrahi öncesi MR veya BT gibi radyolojik yöntemlerle elde edilen bilgiler ışığında bir risiko sınıflaması yapılmaktadır. Örneğin bu tip bir görüntü yönlendirmeli risiko faktörü (Image Defined Risk Factor-IDRF) büyük damarların tümör tarafından çevrelenmesidir. Tümörün cerrahi olarak çıkarılabilirliği olasılığı, hastanın yaşı, tümörün mikroskopik ve moleküler genetik özellikleri evreleme yapılırken dikkate alınır.
Aşağıda her iki evreleme sistemini paralel olarak sunuyoruz. INSS evrelemesi uyarınca lokal hastalık 1-3 arasında evrelendirilirken, metastaz yapmış hastalık evre 4, metastaz yapmış bebeklik nöroblastomu evre 4S olarak sınıflandırılır. INRG evreleme sisteminde lokal sınırlı tümörler belirli risiko faktörlerine bağlı olarak L1 veya L2 olarak, ilerlemiş (metastazlı) hastalık M veya MS olarak sınıflandırılır (ekteki tabloya bakınız).
|
Hastalık evreleri INSS’e göre |
Tanım |
Hastalık evreleri INRG’ye göre |
Tanım |
|---|---|---|---|
|
1 |
Tamamen çıkarılabilmiş tümör |
L1 |
Sadece bir vücut boşluğuna sınırlı, radyolojik olarak risk faktörü bulunmayan (IDRF olmayan) lokal tümör |
|
2 a |
Tamamen çıkarılamamış tümör Hastalık omurga çizgisinde vücudun sadece bir yarımında (orta hattı geçmemiş) Tümörün çevresindeki lenf bezlerind e tutulum yok |
||
|
2 b |
Tamamen çıkarılmış veya çıkarılamamış tümör Hastalık omurga çizgisinde vücudun sadece bir yarımında (orta hattı geçmemiş) Tümörün komşuluğundaki lenf bezlerinde de tutulum var |
L2 |
Bir veya daha çok radyolojik risk faktörü bulunan (IDRF olan) lokalize tümör |
|
3 |
Tümör tamemen çıkarılamamış ve orta hattı geçiyor veya tümörün karşı tarafındaki lenf bezlerinde de tutulum var |
||
|
4 |
Uzak metastaz var (örneğin kemikiliğinde, kemiklerde, karaciğerde, ciltte, çıkarılan lenf bezlerinde veya diğer organlarda) |
M |
MS evresi dışında uzak metastazı olan vakalar |
|
4 S |
Sadece tanı anında 1 yaşından küçük bebeklerde: deri, karaciğer ve/veya minimal olarak kemik iliğine sınırlı metastazlı lokal tümör (evre 1, 2A veya 2B) |
MS |
Metastatik hastalığı sadece deri, karaciğer ve kemikiliğinde olan ve yaşı 18 ayın altında olan hastalar |
Evre 4S ve MS grubundaki hastalar hariç, genel olarak hastalığı daha az ilerlemiş olan hastaların prognozu hastalığı ilerlemiş olan (bu gruba evre 3 ve 4 hastalar girmektedir) hastalara kıyasla daha iyidir. Iyileşme şansı daha az görünen hastalar genel olarak iyileşme şansı yüksek olan hastalara göre çok daha yoğun tedavilere ihtiyaç duyarlar (bakınız tedavi bölümü).
Hastalık seyri
Nöroblastom hastalığının seyri hastadan hastaya değişiklik gösterir. Hastalık seyri özellikle tümörün büyüme şekline ve tanı anında hangi hastalık evresinde olduğuna bağlıdır. Bazı hallerde nöroblastom tanı anında kendi bulunduğu yerle sınırlı olabilir, öte yandan etrafındaki dokulara ve lenf düğümlerine de sıçramış olabilir ve hatta daha uzaktaki organlara da yerleşmiş olabilir (ayrıca yukarıdaki ‘hastalık evreleri’ bölümüne de bakınız). Biyolojik ve klinik olarak iyi seyirli nöroblastomların bir özelliği spontan iyi huylu tümöre değişim gösterebilme (diferansiye olabilme) veya tamamen kaybolabilme (spontan remisyon) potansiyeline sahip olmalarıdır.
Tümör büyümesi ve metastaz oluşturma
Özellikle 18 aydan büyük çocuklarda nöroblastomlar çab uk ve kontrolsüz büyür ve genellikle kan dolaşım sistemi üzerinden veya bazı durumlarda lenf sistemi [lenfatik sistem] üzerinden bütün vücuda dağılabilirler. Bunun sonucunda özellikle kemik iliğinde (hastaların %90’nında), kemiklerde (hastaların %60’ında), çıkartılan lenf düğümlerinde (hastaların %20’si), karaciğerde (hastaların %17’si), daha nadiren de beyinde (%9), deride (%2) ve akciğerde (%1) kardeş tümörler (metastazlar) meydana gelir. Bu durumda evre 4 veya evre M söz konusudur.
Tümörün olgunlaşması
Bazı nöroblastomlar ya spontan olarak yani kendiliklerinden veya bir kemoterapi etkisiyle olgunlaşabilirler ve dolayısıyla daha az kötü huylu tümör hücresi parçaları oluşturabilirler. Bu sürece, tümör olgunlaşması veya ayrışması (diferasyonu) denir. Bu cins tümörler „Ganglio nöroblastom“ diye adlandırılırlar. Gerçi bunlarda hala kötü huylu hücreler bulunmaktadır, ama asıl kötü huylu nöroblastomlardan belirgin derecede daha yavaş bir büyüme gösterirler. Bu tip bir spontan tümör olgunlaşması bazı hastalarda bir yaşından sonra görülebilir. Tamamen olgunlaşma göstermiş ganglionöromlara ancak 4 yaşından sonra veya erişkin hastalarda rastlanır.
Tümörün gerilemesi (spontan regresyon)
Öte yandan kendiliklerinden spontan olarak küçülüp kaybolan nöroblastomlar da vardır. Bu olaya, tümör regresyonu (tümörün gerilemesi) denir. Bu durumda tümör hücreleri adeta kendi kendilerini öldürürler. Uzmanlar buna apopitoz derler.
Tümörün bu şekilde spontan kendiliğinden küçülüp yok olması haline sadece süt çocukluğu döneminde görülen ve evre 4S / evre MS olarak tanımlanan nöroblastomlarda rastlanılır. Bu hastalarda sıklıkla artan metastazlar sebebiyle karaciğer büyümesi görülür; bu bulgu, doğru teşhisin konulmasına yardımcı olur. Oluşan metastazlar önce çabucak büyüyebilirler; bu büyüme esnasında karındaki organları ve akciğeri sıkıştırırlar ve nihayet bu suretle hayati tehlike oluşturabilirler. Bundan sonra kendiliklerinden veya düşük doz bir kemoterapi uygulaması sonrasında küçülüp kaybolabilirler. Spontan tümör regresyonu, sadece süt çocukluğu dönemindeki evre 4S (MS) tümörlerde değil, yaşı daha büyük ve tümör evresi 1-3, L1/L2 olan nöroblastom hastalarında görülebilir.
Nöroblastomların hastalık evreleri ile ilgili detaylı bilgileri tedavi planlama bölümünde bulabilirsiniz.
Tedavi
Nöroblastomlu çocukların tedavisi çocuk onkoloji merkezileri tarafından yapılmalıdır. Ancak bu merkezlerde nöroblastom tedavisi konusunda tecrübeli doctor ve sağlık personeli bulunmaktadır ve bu merkezler modern tedavi yöntemleri hakkında bilgi ve deneyim sahibidir. Ayrıca bu merkezlerde çalışan doktorlar nöroblastom konusunda çalışan diğer merkezlerle sürekli iletişim halindedirler ve hastalarını sıklıkla güncellenen, birlikte oluşturdukalrı tedavi planlarına göre tedavi ederler.
Tedavinin hedefi çocuğu tamamen sağlığına kavuştururken olası erken ve geç yan etkileri olabildiğince aza indirgemektir.
Tedavi yöntemleri
Bir nöroblastom hastasının tedavisi hastanın bireysel hastalık evresine ve hastalık tekrarlama riskine göre düzenlenir. Bazı hastalarda sadece tümörün cerrahi olarak çıkarılması veya tümörden sadece biyopsi örneğinin alınması yeterli olabilirken, diğer hastalarda iyileşme oranlarının arttırılabilmesi için çeşitli tedavi yöntemlerinin birlikte kullanılması gerekmektedir.
Burada anlatılan tedavi yöntemleri Almanya'daki mevcut tedavi standardını temsil etmektedir. Klinik çalışmaların bir parçası olarak daha yeni tedavi kavramları sürekli olarak gözden geçirilmektedir, bu nedenle bazı hastalarda bireysel tedavi bu standartlardan sapabilir.
Nöroblastomda kullanılabilecek tedavi yöntemleri cerrahi (operasyon), kemoterapi ve radyoterapidir (ışın tedavisi). Hastalığı tekrarlama riski çok yüksek hastalarda bunlara ek olarak yüksek doz kemoterapiyi takiben otolog kök hücre nakli ve antikorlar ile immunoterapi de uygulanmaktadır. Bu hastalarda tamamlayıcı olarak radyoaktif işaretli metiliyotbenzilguanidin tedavisi (MIBG tedavisi) de kullanılabilmektedir.
Operasyonun (ameliyat, cerrahi girişimin) amacı tümörün tamamen çıkarılması ve/veya tümörden doku örneği alınabilmesidir. Kemoterapide [kemoterapi] hücre bölünmesini engelleyen (sitostatik) ve böylece hücrenin ölmesini sağlayan ilaçlar kullanılır. Genel olarakkötü huylu hücrelere mümkün olan en büyük zararın verilebilmesi için farklı şekilde etki mekanizmaları olan ilaçlar bir arada kulanılır (polikemoterapi). Daha yoğun bir tedavi ise yüksek doz kemoterapidir: bu tedavi sadece kanser hücrelerini değil, aynı zamanda kemikiliğindeki kan yapıcı hücreleri [kan kök hücreleri] de öldürür, bu nedenle bu tedaviyi takiben otolog kök hücre nakli yapılması gerekir. Yüksek riskli hastalarda gerekli olan radyasyon tedavisi, dışarıdan cilt yoluyla etkilenen bölgeye yayılan yüksek enerjili, elektromanyetik radyasyon (perkütan radyasyon tedavisi, radyoterapi) ile gerçekleştirilir. Böylece tümör hücrelerinin üreme özellikleri zarara uğratılarak ölmeleri sağlanır.
Yukarıda sayılan yöntemlerin hangisinin ve hangi kombinasyonla uygulanacağı, özellikle tümörün yayılımına, ameliyat edilebilirlik durumuna, habislik derecesine ve hastanın yaşına bağlıdır (tedavi planlaması bölümüne bakınız). Hastalık ne kadar çok ilerlemişse, tümörün saldırganlık ve büyüme riski ne kadar fazlaysa veya tedaviden sonra nüksetme (tekrarlama) tehlikesi ne kadar büyükse, uygulanması gereken tedavide o oranda yoğun ve karmaşık olacaktır.
Nöroblastom tedavisi sırasında çeşitli yan etkiler ortaya çıkabileceği için, tedavi sırasında eş zamanlı olarak bu yan etkilerin ortaya çıkmasını önleyici ve/veya bu yan etkileri azaltan destekleyici (supportif) tedaviler de uygulanır. Şu bölümde destek tedavisi (supportif tedavi) hakkında bilgi bulabilirsiniz.
Tedavi işleyişi
Yukarıdaki sebeplerden ötürü her hasta, tedavinin başında ve/veya bir ameliyat sonrasında ve/veya numune alınmasını takiben, bir risk ve terapi grubuna ayrılır. Güncel terapi yönergeleri bugün için üç terapi grubu öngörmektedir: Gözlem grubu, orta risk grubu ve yüksek risk grubu. Bu terapi gruplarının her birine özgü değişik terapi planları geçerlidir. Bu suretle her hastaya özgü ve risk adaptasyonu saptanmış bir tedavi gerçekleştirilir.
Düşük rik grubundaki hastalarda tedavi (gözlem grubu)
Lokalize (bölgesel) tümörü olan ve/veya yaşı nedeni ile tedavi stratejisi olarak beklemenin hastaya zarar vermeyeceği gruptaki hastalar düşük risk grubuna (gözlem grubu) alınır. Bir hastanın gözlem grubuna alınabilmesi için kötü moleküler genetik özelliklere (örneğin MYCN amplifikasyonu veya 1 p delesyon gibi) sahip olmaması gerekir. Gözlem grubuna alınacak hasta grupları şunlardır:
- Evre 1 (INSS), yaş 0-21 ay, MYCN amplifikasyonu yok
- Evre 2 (INSS), yaş 0-21 ay, hem MYCN amplifikasyonu hem de 1p delesyonu yok
- Evre 3 (INSS), yaş 0-2 hem MYCN amplifikasyonu hem de 1p delesyonu yok
- Evre 4S (INSS), INRG evre MS önerisi uyarınca yaşı 0-18 ay arası olan, hem MYCN amplifikasyonu hem de 1p delesyonu olmayan hastalar
Gözlem grubu: Düşük riskli hastalarda, spontan tümör gerileme oranının yüksek olması sebebi ile tedavi sadece tümörün cerrahi olarak tamamen çıkarılması; hatta sadece doku örneği alınması (biyopsi) ile sınırlıdır. Genel durumu iyi olan hastalarda kemoterapiye veya ışın tedavisine gerek yoktur. Ancak hastalar düzenli klinik muayene, ultrasonografi, manyetik rezonans görüntüleme, tümör belirteçleri gibi yöntemlerle çok yakından izlenirler. İlk yıl içinde hastalar en az 6 haftada bir; 2-5.inci yıllar arasında en az 3 ayda bir, daha sonra da en az 6 ayda bir veya yılda bir incelenirler. Seçilecek izlem yöntemi hastada tümör kalıp kalmamış olmasına ve tümörün ultrason ile iyi gösterilebilecek bir bölgede olup olmamasına göre değişiklik gösterebilir.
Eğer geriye kalan tümör cerrahi sonrası ilk 12 içinde yeniden ortaya çıkarsa veya büyümeye devam ederse veya teadviye ihtiyaç gösteren semptomlara neden olursa (örneğin hastanın genel durumunun kötü olması, beslenme problemleri, kilo kaybı, yüksek kan, idrar akış problemleri gibi), tümörün gerilemesini sağlamak için çok hafif bir kemoterapi uygulanır. Tedavi doksorubisin, vinkristin, siklofosfamid kombinasyonundan oluşan 4 kemoterapi bloğu olarak uygulanabilir. Alternatif olarak karboplatin ve etoposid de kullanılabilir. Tümör büyümesi sağlanır sağlanmaz kemoterapiye son verilir. Bazı hastalarda akabinde tümörün tamamen çıkarılması veya semptomların azaltılması için ek bir cerrahi girişim söz konusu olabilir. Bu durum özellikle evre 4S olan hastalarda tümör küçülmeye başlamadan önce hızla büyüyebileceği için söz konusu olabilir.
Orta risk grubunda tedavi
Hastalığı daha ileri evrede olan ve/veya yaşları büyük olan ve/veya kötü prognostik moleküler genetik bulguları olan (örneğin 1p delesyonu) hastalar orta risk grubunda tedavi alırlar. Hastanın orta risk grubunda olması için MYCN amplifikasyonuna sahip olmaması ön koşuldur.
- Evre 2 (INSS), yaş 0-21, 1 p delseyonu olan ama MYCN amplifikasyonu olmayan hastalar
- Evre 3 (INSS), yaş 0-21, 1 p delseyonu olan ama MYCN amplifikasyonu olmayan hastalar
- Evre 3 (INSS), yaş 2-21, hem 1 p delesyonu hem de MYCN amplifikasyonu olmayan hastalar
- Evre 4 (INSS), INRG evre MS önerisi uyarınca yaşı 0-18 ay arası olan, MYCN amplifikasyonu olmayan hastalar
Tedavi süreci: Tedavi için cerrahi uygulanır, cerrahi (amelyiat) mümkün değilse öncelşkle biyopsi yapılır. Cerrahiyi takiben kemoterapi uygulanır. Kemoterapi, altı adet yoğun kemoterapi (indüksiyon tedavisi, hücum tedavisi) bloğunu takiben 4 tane nispeten hafif idame tedavisinden oluşur. Kemoterapi öncesi cerrahi uygulanamadı ve sadece biyopsi alındı ise, ilk bir kaç hücum tedavisi sonrası, sıklıkla tedavi ile tümör küçülmüş olacağı için, tümörün cerrahi olarak çıkarılması hedeflenir.
Hücum tedavisi çerçevesinde doksorubisin, vinkristin, siklofosfamid veya karboplatin, etoposid, vindesin kombinasyonundan oluşan kemoterapi blokları dönüşümlü olarak uygulanır. İlaçlar saatler veya günler süren infüzyonlar şeklinde verilir. İdame tedavisinde tablet şekilnde siklofosfamid kullanılır.
Yoğun kemoterapi sonrasında hala aktif tümör tespit edilirse, 18 aydan büyük çocuklarda idame tedavisi ile eş zamanlı olarak tumor bilgesi ışınlanır (36-40 Gy ışın dozu kullanılarak). Daha önceden kalmış olabilen kalıntı bir tümör (örneğin ilk başta cerrahi değil sadece biyopsi yapıldıysa) tedavi sırasında veya tedaviyi takiben yeni bir cerrahi girişim ile tamamen çıkarılabilir. Tüm tedavi süresi yaklaşık bir yılı bulur.
Yüksek risk grubunda tedavi
Diğer gruplara girmeyen ve 18 ay üstündeki tüm evre 4 hastalar yüksek risk grubunda olarak değerlendirilir. Yüksek riskli hastalar için tedavi konsepti çok geniş kapsamlıdır.
Tedavi süreci: Bu gruptaki hastalara tümörün tamamen çıkarılması veya biyopsi sonrası yaklaşık 5 ay süren, çeşitli ilaç kombinasyonlarından oluşan, hücum tedavisi (indüksiyon tedavisi) de denilen yoğun kemoterapi blokları uygulanır. Günümüzdeki standart hücum tedavisi dönüşümlü olarak kullanılan sisplatin/etoposid/vindesin ve vinkristin/dakarbazin/ifosfamid/doksorubisin kombinasyonlarından oluşan 6 bloğu içerir. Kemoterapi blokları arasında veya bloklar sonrasında tümörün tamamen çıkarılmasını hedefleyen ikinci cerrahi girişim uygulanır. Bundan sonra tüm hastalara yüksek doz kemoterapiyi takiben otolog kök hücre nakli uygulanır (yaklaşık 6 hafta sürer). MIBG positif kalıntı tümörü olan hastalarda yüksek doz kemoterapi ile birlikte radyoaktif işaretli metiliyotbenziguanidin ile 131-I-MIBG tedavisi uygulanabilir. Bu durumda I-MIBG tedavisi yüksek doz kemoterapiden önce yapılır.
Yüksek doz tedavinin ardından tümör yatağı ışınlanır ve Dinutuksimab beta isimli antikor ile immunoterapi uygulanır. Yüksek doz tedaviyi takiben tümör yatağı ışınlanır ve dinutuksimab adı verilen antikor ile immunoterapi uygulanır. Bu tedavi döneminin amacı (idame tedavisi veya pekiştirme tedavisi sonrası tedavi de denir) geriye kalmış olabilecek tümör hücrelerini yok etmektir. Eğer aktif bir tümör söz konusu ise ışınlama dozu 36 gy olarak önerilmektedir. Tüm tedavi süresi 2 yılı bulabilir.
Tedavi iyileştirme araştırmaları ve veri bankası
Almanya’da hemen hemen tüm nöroblastomlu çocuk ve gençler tedavi iyileştirme araştırmaları veya veri tabanları çerçevesinde tedavi edilirler.
Tedavi iyileştirme çalışmaları hasta çocukların mevcut en güncek bilimsel veriler ışığında tedavi edilmelerini sağlayan ve tedavi seçeneklerini sürekli iyileştiren ve geliştiren kontrollü klinik çalışmalardır. Tanı sırasında bir çalışma mevcut olmadığı için veya çalışmaya alınma kriterlerine uymadıkları için herhangi bir tedavi iyileştirme çalışma protokolüne alınamayan hastalar sıklıkla veri tabanlarında toplanırlar. Bu veri tabanlarının amacı hastaların tedavilerine bilimsel olarak eşlik etmektir. Veri tabanları sayesinde, veri tabanının yürütücüsü çalışma grubu hastayı takip eden doktorlara detaylı tedavi önerilerinde bulunurlar; böylece hastanın en ideal tedaviyi alması sağlanmış olur.
İkibinonaltı yılının sonuna kadar yeni tanı almış süt çocukları, küçük çocuklar ve gençler için 2 tedavi iyileştirme çalışması bulunmaktaydı: Gözlem grubunda ve/veya orta risk grubundaki hastalar için NB2004 protokolü; yüksek risk grubundaki hastalar için NB 2004-HR protokolü. Her iki çalışmada şu an sonlanmış bulunmaktadır, çalışmaların sonuçları toparlanma aşamasındadır. 2022 yılı içinde yeni devam çalışmalarının açılması planlanmaktadır.
Halen yeni tanı alan nöroblastom hastaları ve nöroblastom hastalığı tekrarlayan vakalar NB Register 2016 isimli nöroblastom veri bankasına kaydedilebilirler (aşağıya bakınız). Hastalığı nüks eden (tekrarlayan) veya tedaviye yanıt vermeyen yüksek riskli hastalar için Berlin, Köln ve Greifswald’daki çalışma merkezlerinden faz I/II çalışmalar hakkında bilgi alınabilir.
NB 2016 veri tabanı/bankası (NB Register 2016)
01.01.2017 tarihinden beri yeni nöroblastom tanısı alan (nöroblastom, ganglionöroblastom, ganglionörom) yenidoğan, süt çocuğu, çocuk ve gençler (hatta erişkinler) için veya hastalığı tekrar eden (nüks eden) hastalar için „NB Register 2016“ (NB 2016 veri tabanı) veri tabanına kaydolma imkanı bulunmaktadır.
Bu veri tabanının amacı hastalığın sıklığı, genel seyri ve hastalığın uzun dönem yan etkileri hakkında bilgi edinmek ve hastalığın seyrini (prognozunu) iyileştirmektir. Tedavi seçimini, çalışma merkezinden aldığı tedavi önerileri doğrultusunda tedaviyi yürüten hekim yapar. Veri tabanına kayıtlı olmak ileride açılabilecek bir tedavi iyileştirme çalışmasına katılmaya engel oluşturmaz.
Veri tabanı yönetimini Köln Üniversite kliniğinden Prof. Dr. med. Thorsten Simon yapmaktadır. Veri tabanı hakkındaki bilgilere; Berlin, Köln ve Greifswald’daki çalışma merkezlerine şu linkten ulaşabilirsiniz.
Tedavi başarısı (Prognoz)
Bir nöroblastom hastasının iyileşip iyileşmeyeceği konusunda birşey söylemek çok zordur ve hastadan hastaya değişkenlik göstermektedir. Bu konuda hastalığın evresi, tümörün saldırganlığı ve hastanın yaşı gibi faktörler rol oynamaktadır. Nöroblastom hastalığı 4S evresindeki çocuklarda, genellikle bölgesel sınırlı tümörlerde ve daha küçük çocuklarda veya hastalığı evre 1-2 olan lokalize hastalıklı çocuklarda çok iyi bir prognoz (10 yıllık sağkalım %90’dan fazla) söz konusudur. Molekular genetik olarak yüksek risk kriteri olmayan 18 ayın altındaki evre 3. Yüksek risk kriteri olamayan küç genellikle bölgesel sınırlı tümörlerde ve daha küçük çocuklarda iyi bir prognoz mümkün olabilmektedir. Metastaz oluşturmuş nöroblastomlu 4 büyük çocuklarda ise iyileşme beklentileri, uygulanan yoğun tedaviye rağmen hala olumsuzdur.
Uyarı: Yukarıda sözü edilen iyileşme oranları istatistiksel verilerdir. Yalnızca tüm Wilms tümörlü hastalar için önemli ve gerçeğe uygun bir ifade oluşturmaktadır. Bir hastanın iyileşeceği veya iyileşmeyeceği konusunda istatistiğe dayanarak bir şey söylemek mümkün değildir.
Diğer bilgiler
Burada verilen bilgiler nöroblastomlu çocuk ve gençlerde uygulanan tedaviler ile ilgili aşağıdaki bilimsel yayınlar, güncel tedavi kuralları ve şemaları göz önüne alınarak nöroblastom tedavi merkezi ile birlikte yapılan ortak çalışma sonucunda oluşturulmuştur. Ek sorularınız için her zaman tedavinizi yapmakta olan hekime başvurabilirsiniz.
Kaynakça 
- Erdmann F, Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Annual Report 2019 (1980-2018). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2020 [URI: https://www.kinderkrebsregister.de/ typo3temp/ secure_downloads/ 42507/ 0/ 1c5976c2ab8af5b6b388149df7182582a4cd6a39/ Buch_DKKR_Jahresbericht_2019_komplett.pdf]
- Simon T: Leitlinie: Neuroblastom. S1-Leitlinie 025-008 (Leitlinie der Gesellschaft für Pädiatrische Onkologie und Hämatologie) AWMF-online 2019 [URI: https://www.awmf.org/ uploads/ tx_szleitlinien/ 025-008l_S1_Neuroblastom_2019-07_01.pdf]
- Eggert A, Simon T, Hero B, Lode H, Ladenstein R, Fischer M, Berthold F: Neuroblastom. in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie Springer Verlag GmbH GDeutschland 2006, 2018, 2. vollständig überarbeitete Auflage 2018, 420 [ISBN: 978-3-662-43685-1]
- Berthold F, Spix C, Kaatsch P, Lampert F: Incidence, Survival, and Treatment of Localized and Metastatic Neuroblastoma in Germany 1979-2015. Paediatric drugs 2017, 19: 577 [PMID: 28786082]
- Fischer J, Pohl A, Volland R, Hero B, Dübbers M, Cernaianu G, Berthold F, von Schweinitz D, Simon T: Complete surgical resection improves outcome in INRG high-risk patients with localized neuroblastoma older than 18Â months. BMC cancer 2017 Aug 4; 17: 520 [PMID: 28778185]
- Simon T, Hero B, Schulte JH, Deubzer H, Hundsdoerfer P, von Schweinitz D, Fuchs J, Schmidt M, Prasad V, Krug B, Timmermann B, Leuschner I, Fischer M, Langer T, Astrahantseff K, Berthold F, Lode H, Eggert A: 2017 GPOH Guidelines for Diagnosis and Treatment of Patients with Neuroblastic Tumors. Klinische Padiatrie 2017, 229: 147 [PMID: 28561228]
- Oberthuer A, Berthold F, Hero B, Till H: Neuroblastome, in: Solide Tumoren im Kindesalter. Fuchs J (Hrsg.) Schattauer GmbH: Stuttgart 2012, 77 [ISBN: 978-3-7945-2786-1]
- Brisse HJ, McCarville MB, Granata C, Krug KB, Wootton-Gorges SL, Kanegawa K, Giammarile F, Schmidt M, Shulkin BL, Matthay KK, Lewington VJ, Sarnacki S, Hero B, Kaneko M, London WB, Pearson AD, Cohn SL, Monclair T, International Neuroblastoma Risk Group Project: Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology 2011, 261: 243 [PMID: 21586679]
- Øra I, Eggert A: Progress in treatment and risk stratification of neuroblastoma: impact on future clinical and basic research. Seminars in cancer biology 2011, 21: 217 [PMID: 21798350]
- Hero B, Papenheim H, Schuster U: Neuroblastom – Informationen für Eltern. Fördergesellschaft Kinderkrebs-Neuroblastom-Forschung e.V., Baden 2011 [URI: http://www.neuroblastoma.de/ fileadmin/ PDF/ Neuroblastom.pdf]
- Maris JM: Recent advances in neuroblastoma. The New England journal of medicine 2010 Jun 10; 362: 2202 [PMID: 20558371]
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, von Schweinitz D, Simon T, Cohn SL, Pearson AD, INRG Task Force: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. Journal of clinical oncology 2009, 27: 298 [PMID: 19047290]
- Oberthuer A, Theissen J, Westermann F, Hero B, Fischer M: Molecular characterization and classification of neuroblastoma. Future oncology (London, England) 2009, 5: 625 [PMID: 19519203]
- Fischer M, Spitz R, Oberthür A, Westermann F, Berthold F: Risk estimation of neuroblastoma patients using molecular markers. Klinische Padiatrie 2008, 220: 137 [PMID: 18478485]
- Hero B, Simon T, Spitz R, Ernestus K, Gnekow AK, Scheel-Walter HG, Schwabe D, Schilling FH, Benz-Bohm G, Berthold F: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. Journal of clinical oncology 2008, 26: 1504 [PMID: 18349403]
- Ebell W: Hämatopoetische Stammzelltransplantation. in: Gadner H, Gaedicke G, Niemeyer CH, Ritter J:. Pädiatrische Hämatologie und Onkologie Springer-Verlag, 2006, 66 [ISBN: 3540037020]
- Claviez A, Lakomek M, Ritter J, Suttorp M, Kremens B, Dickerhoff R, Harms D, Berthold F, Hero B: Low occurrence of familial neuroblastomas and ganglioneuromas in five consecutive GPOH neuroblastoma treatment studies. European journal of cancer (Oxford, England : 1990) 2004, 40: 2760 [PMID: 15648116]
- Berthold F, Hero B, Kremens B, Handgretinger R, Henze G, Schilling FH, Schrappe M, Simon T, Spix C: Long-term results and risk profiles of patients in five consecutive trials (1979-1997) with stage 4 neuroblastoma over 1 year of age. Cancer letters 2003, 197(1-2): 11 [PMID: 12880954]
- Hero B, Berthold F: Neuroblastom. Monatschr Kinderheilkd 2002, 150: 775 [DOI: 10.1007/s00112-002-0493-0]
 Nöroblastom (Kısa Bilgiler) (497KB)
Nöroblastom (Kısa Bilgiler) (497KB)