Neuroblastom – Kurzinformation
Das Neuroblastom ist eine bösartige Erkrankung des sympathischen Nervensystems, die vor allem im frühen Kindesalter auftritt. In diesem Text erhalten Sie die wichtigsten Informationen zu Krankheitsbild und Krankheitsverläufen sowie zu Häufigkeit, möglichen Ursachen, Symptomen, Diagnose, Therapieplanung, Behandlung und Prognose der Erkrankung.
Autor: Maria Yiallouros, Redaktion: Maria Yiallouros, Freigabe: Prof. Dr. med. Frank Berthold, Prof. Dr. med. Angelika Eggert, Prof. Dr. med. Thorsten Simon, Zuletzt geändert: 08.04.2025 https://dx.doi.org/10.1591/poh.neurobl.patinfo.kurz.1.20120611
Inhaltsverzeichnis
Krankheitsbild
Neuroblastome sind bösartige solide Tumoren. Sie entstehen aus entarteten unreifen Zellen des sympathischen Nervensystems [sympathisches Nervensystem], welches Teil des autonomen (vegetativen) Nervensystems ist [siehe autonomes Nervensystem].
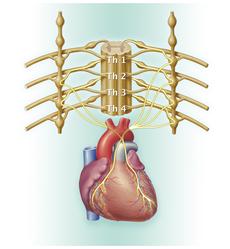
Neuroblastome können überall dort auftreten, wo sich sympathisches Nervengewebe befindet. Am häufigsten entstehen sie im Nebennierenmark (etwa 50 %) und im Bereich der Nervengeflechte beidseits der Wirbelsäule, im so genannten Grenzstrang. Wenn der Grenzstrang betroffen ist, können Neuroblastome auf jeder Höhe entlang der Wirbelsäule vorkommen: im Bauch-, Becken-, Brust- und Halsbereich. In der Mehrzahl der Fälle (zu etwa 75 %) befindet sich der Tumor im Bauchraum, etwa ein Fünftel der Tumoren liegen im Brust- und Halsbereich.
Manche Neuroblastome sind auf den Ursprungsort begrenzt, andere streuen in nahe gelegene Lymphknoten. Bei etwa der Hälfte der Patienten findet man zum Zeitpunkt der Diagnose auch Absiedelungen der bösartigen Zellen (Metastasen) in Knochenmark, Knochen, entfernten Lymphknoten oder in der Leber, seltener in Gehirn, Lunge oder der Haut. Eine Besonderheit biologisch günstiger Neuroblastome ist, dass sie sich spontan zurückbilden können (siehe auch Abschnitt "Krankheitsverläufe").
Häufigkeit
Neuroblastome machen etwa 5,5 % aller Krebserkrankungen im Kindes- und Jugendalter aus. Sie gehören nach den Tumoren des Zentralnervensystems (ZNS-Tumoren, Hirntumoren) zu den häufigsten soliden Tumoren in dieser Altersgruppe. In Deutschland erkranken nach Angaben des Deutschen Kinderkrebsregisters (Mainz) jährlich etwa 120 Kinder und Jugendliche unter 18 Jahren neu an einem Neuroblastom. Damit sind pro Jahr etwa 11 von 1.000.000 Kindern unter 18 Jahren von dieser Krankheit betroffen.
Da Neuroblastome embryonale Tumoren sind, kommen sie vor allem im frühen Kindesalter vor: 90 % der Patienten sind jünger als sechs Jahre alt. Am häufigsten betroffen sind, mit etwa 46 %, Neugeborene und Säuglinge im ersten Lebensjahr. Der Altersdurchschnitt bei der Diagnose liegt bei 14 Monaten. Jungen erkranken etwa 40 % häufiger als Mädchen (Geschlechterverhältnis 1,4 : 1). Ein Neuroblastom kann aber auch bei älteren Kindern, Jugendlichen und im Einzelfall sogar bei Erwachsenen vorkommen.
Ursachen
Die Ursachen für die Entstehung eines Neuroblastoms sind noch weitgehend ungeklärt. Bekannt ist, dass die Krankheit durch eine bösartige Veränderung (Entartung) von unreifen Zellen des sympathischen Nervensystems ausgelöst wird. Die Fehlentwicklung dieser noch nicht ausgereiften (embryonalen) Nervenzellen beginnt möglicherweise bereits vor der Geburt und kann eine Folge von Chromosomenveränderungen und/oder Genveränderungen (Mutationen) sein.
Verschiedene genetische Veränderung wurden in Neuroblastomzellen bereits nachgewiesen, jedoch sind diese sehr heterogen, das heißt, es lässt sich keine spezifische Erbgutveränderung beobachten, die konstant in allen Tumoren auftritt. Insgesamt sind vermutlich eine Reihe genetischer und auch epigenetischer Veränderungen an der Entstehung eines Neuroblastoms beteiligt. Eine Vererbung im eigentlichen Sinne liegt nach dem derzeitigen Kenntnisstand der Forschung bei den meisten Patienten nicht vor.
Es gibt allerdings Familien, in denen Neuroblastome und verwandte Tumoren über mehrere Generationen gehäuft auftreten. Etwa 1-2 % der Patienten sind davon betroffen, häufig haben sie mehr als nur einen Primärtumor. Darüber hinaus können Neuroblastome auch in Verbindung mit einem so genannten Krebsprädispositionssyndrom auftreten, einer genetisch bedingten Erkrankung, die unter anderem mit einer erblichen Veranlagung für Tumoren einhergeht. Krebsprädispositionssyndrome, die bei der Entstehung eines Neuroblastoms eine Rolle spielen können, sind zum Beispiel der Morbus Hirschsprung oder das Undine-Syndrom.
Bei der überwiegenden Mehrzahl der Patienten entsteht die Erkrankung jedoch durch spontane Mutationen oder andere genomische Veränderungen im Erbgut von Körperzellen. Ob auch äußere Einflüsse (wie Umweltfaktoren, berufliche Belastung der Eltern, Medikamenteneinnahme, Nikotin- oder Alkoholkonsum während der Schwangerschaft) eine Rolle spielen können, ist bislang nicht erwiesen.
Krankheitszeichen
Viele Patienten mit Neuroblastom haben keine Krankheitszeichen (Symptome). Bei ihnen wird der Tumor zufällig entdeckt, zum Beispiel bei einer Routineuntersuchung durch den Kinderarzt oder bei einer Ultraschall- oder Röntgenuntersuchung, die aus einem anderen Anlass durchgeführt wird. Beschwerden treten in der Regel erst dann auf, wenn das Tumorwachstum fortgeschritten ist, Tochtergeschwülste (Metastasen) auftreten oder umgebende Strukturen beeinträchtigt sind.
Darüber hinaus sind die Krankheitszeichen vielfältig. Sie variieren je nach Lage des Tumors oder der Metastasen. Tastbare Tumoren oder Metastasen können erste Symptome sein, bei manchen Kindern fällt eine Schwellung am Bauch oder am Hals auf. Tumoren des Bauchraumes oder der Nebenniere können unspezifische Symptome wie Bauchschmerzen, Verstopfung, Völlegefühl oder Durchfall verursachen; durch Druck auf den Harnleiter kann es auch zu einem Harnstau kommen. Befindet sich der Tumor im Brustraum, kann der Druck auf die Lunge zu Husten, Lungenentzündung oder Luftnot führen. Wirbelsäulennahe Tumoren (Tumoren des Grenzstrangs) können in den Wirbelsäulenkanal einwachsen und dadurch neurologische Symptome – wie Nervenschmerzen, Blasen- oder Darmentleerungsstörungen oder gar Lähmungserscheinungen – verursachen.
Bluthochdruck oder anhaltende Durchfälle können in seltenen Fällen durch die hormonelle Aktivität des Tumors entstehen. Bei Tumoren im Halsbereich oder oberen Brustbereich kann das so genannte Horner-Syndrom auftreten. Darunter versteht man ein Zurücksinken des Augapfels mit einseitig verkleinerter Pupille und hängendem Lid. Weitere Veränderungen im Bereich der Augen können Lidekchymosen und, bei fortgeschrittener Erkrankung, manchmal Blutergüsse um die Augen sein (so genanntes Brillenhämatom, Monokelhämatom). Eine seltene Verlaufsform ist das Neuroblastom mit Opsomyoklonus-Ataxie-Syndrom (OMAS).
Knochenmetastasen – die vor allem die langen Röhrenknochen von Armen und Beinen sowie die Knochen des Schädels und der Augenhöhle betreffen – können zu Knochenschmerzen führen. Bei manchen Patienten äußern sich Knochenschmerzen durch einen humpelnden Gang. Wenn das Knochenmark stark infiltriert ist, können Blutarmut (Anämie), ein Mangel an Blutplättchen (Thrombozytopenie) sowie weißen Blutzellen (Leukopenie) mit entsprechender Infekt- und Blutungsneigung auftreten.
Allgemeine Symptome, die auf ein, häufig fortgeschrittenes, Neuroblastom hinweisen können, sind:
- Müdigkeit, Lustlosigkeit, Leistungseinschränkung, Schwäche, Blässe
- anhaltendes mäßiges Fieber ohne erkennbare Ursache, Schwitzen
- Knoten oder Schwellungen an Bauch oder Hals; Lymphknotenschwellungen
- aufgetriebener, großer Bauch
- Verstopfung oder Durchfälle, Bauchkoliken
- Appetitlosigkeit, Übelkeit, Erbrechen; infolgedessen Gewichtsverlust
- Knochenschmerzen
- Brillenhämatome
Gut zu wissen: Das Auftreten eines oder mehrerer dieser Krankheitszeichen wird allerdings nur selten bedeuten, dass ein Neuroblastom vorliegt. Viele dieser Symptome können auch vergleichsweise harmlose Ursachen haben. Bei Beschwerden ist es jedoch ratsam, so bald wie möglich einen Arzt zu konsultieren, um deren Ursache zu klären.
Diagnose
Findet der (Kinder-)Arzt durch Krankheitsgeschichte (Anamnese) und körperliche Untersuchung Hinweise auf ein Neuroblastom, wird er den Patienten in ein Krankenhaus überweisen, das auf diese Form der Krebserkrankung spezialisiert ist (kinderonkologische Behandlungseinrichtung). Denn bei Verdacht auf ein Neuroblastom sind verschiedene Untersuchungen notwendig, zunächst um die Diagnose zu sichern, dann aber auch um festzustellen, um welche Form des Neuroblastoms es sich handelt und wie weit sich die Erkrankung ausgebreitet hat. Die Klärung dieser Fragen ist Voraussetzung für eine optimale Behandlung und Prognose des Patienten.
Laboruntersuchungen
Neben einer erneuten, eingehenden Anamnese und körperlichen Untersuchung spielen bei der Diagnosestellung zunächst Laboruntersuchungen eine wichtige Rolle. Bei den meisten Patienten mit einem Neuroblastom findet man im Blut oder im Urin erhöhte Werte bestimmter Substanzen, die als "Tumormarker" für die Krankheitsdiagnose (vor allem aber im weiteren Krankheitsverlauf für die Kontrolle des Therapieerfolgs) genutzt werden können. Wichtige Tumormarker beim Neuroblastom sind bestimmte Katecholamine oder deren Abbauprodukte (Dopamin, Vanillinsäure, Homovanillinsäure) sowie die Neuronspezifische Enolase (NSE).
Bildgebende Untersuchungen zum Tumornachweis
Weitere Untersuchungen, die der Sicherung der Diagnose sowie der Abgrenzung eines Neuroblastoms von anderen Erkrankungen (wie Wilms-Tumor, Phäochromozytom) dienen, sind bildgebende Verfahren: Bereits mit Hilfe einer Ultraschalluntersuchung (Sonographie) können Lage und Größe der meisten Neuroblastome sowie ein eventueller Lymphknotenbefall im Halsbereich oder im Bauch- und Beckenraum sehr gut sichtbar gemacht werden. Eine Röntgenaufnahme [siehe Röntgenuntersuchung] dient der Überprüfung von Lunge und Brustraum.
Um auch sehr kleine Tumoren erkennen und die Beziehung zu benachbarten Strukturen (wie Organe, Blutgefäße, Nerven) besser beurteilen zu können, wird zusätzlich eine Magnetresonanztomographie (MRT) mit und ohne Kontrastmittel durchgeführt. In Einzelfällen kann an Stelle der MRT auch eine Computertomographie (CT) in Frage kommen. Prinzipiell wird die MRT aber bevorzugt eingesetzt, da sie, anders als die CT, nicht mit Röntgenstrahlung, sondern mit Magnetfeldern arbeitet und somit keine Strahlenbelastung verursacht.
Untersuchungen zur Metastasensuche
Zum Nachweis beziehungsweise Ausschluss von Metastasen sowie zur weiteren Beurteilung des Primärtumors erfolgt zudem eine Ganzkörperszintigraphie mit der schwach radioaktiv markierten Substanz 123Iod-meta-Iodbenzylguanidin, kurz 123I-mIBG (MIBG-Szintigraphie). Wenn die MIBG-Szintigraphie negativ ausfällt, das heißt, keine Ergebnisse zeigt, können alternativ andere Methoden der Szintigraphie angezeigt sein, zum Beispiel eine Positronen-Emissions-Tomographie (PET) mit radioaktiv markiertem Zucker (18-Fluor-Deoxyglukose, kurz FDG). Beide Verfahren werden mit einer CT oder MRT kombiniert.
Da sich mit Hilfe der Szintigraphie ein sehr geringer Befall des Knochenmarks nicht feststellen lässt, ist bei allen Patienten die Entnahme von Knochenmark notwendig. Das Knochenmark wird mittels Knochenmarkpunktion oder Knochenmarkstanzbiopsie an vier unterschiedlichen Stellen gewonnen, meist in Kurznarkose, und anschließend unter dem Mikroskop und mit Hilfe von Spezialverfahren auf bösartige Zellen untersucht. Bei Patienten mit Metastasen wird auch eine MRT des Schädels durchgeführt, um einen Befall des Gehirns auszuschließen. Auch eine Ganzkörper-MRT kann in solchen fortgeschrittenen Krankheitsstadien in Frage kommen, unter anderem, um mögliche Knochenmetastasen nachzuweisen.
Gewebeentnahme (Biopsie)
Prinzipiell ist die endgültige Sicherung der Diagnose nur durch eine feingewebliche Untersuchung von Tumorgewebe möglich. Die Entnahme von Tumormaterial erfolgt in der Regel mit der Operation. Molekulargenetische Untersuchungen [Molekulargenetik] des entnommenen Gewebes erlauben Rückschlüsse auf das Maß der Bösartigkeit des Tumors. Denn bestimmte Veränderungen (Mutationen) in der Tumor-DNA (wie die so genannte MYCN-Amplifikation oder 1p-Deletion) sowie die Ausprägung unterschiedlicher Genmuster (Fachleute sprechen von einer ungünstigen Genexpressionssignatur) korrelieren mit einer ungünstigen Prognose, während das Fehlen dieser Veränderungen oder andere Mutationen mit einer günstigeren Prognose einhergehen können.
Vor einigen Jahren wurden zusätzliche Gendefekte in Neuroblastomzellen entdeckt (zum Beispiel Veränderungen des ALK-Gens oder die so genannte Telomerase-Aktivierung), die im Falle eines Erkrankungsrückfalls teilweise auch therapeutisch genutzt werden können, sofern sie bei der entsprechenden Neuroblastom-Erkrankung vorliegen.
Behandlungsvorbereitende Untersuchungen
Je nach Art der geplanten Behandlung können vor Therapiebeginn weitere Untersuchungen hinzukommen, um den Zustand und die Funktion bestimmter Organe zu überprüfen. Dazu gehören – insbesondere vor einer Chemotherapie – die Überprüfung der Herzfunktion (Elektrokardiographie [EKG], Echokardiographie), der Hörfunktion (Audiometrie) und der Nierenfunktion, ein Nieren-Ultraschall oder auch eine Röntgenuntersuchung der Hand, welche Aufschluss über das Wachstumsverhalten des Kindes gibt. Veränderungen, die möglicherweise im Laufe der Therapie auftreten, können aufgrund solcher Ausgangsbefunde besser beurteilt und bei der Behandlung entsprechend berücksichtigt werden.
Gut zu wissen: Nicht alle der genannten Untersuchungen sind bei jedem Patienten notwendig. Andererseits können eventuell Untersuchungen hinzukommen, die hier nicht erwähnt wurden. Fragen Sie Ihren behandelnden Arzt oder das Behandlungsteam, welche Untersuchungen bei Ihrem Kind geplant sind und warum die jeweilige Untersuchung erforderlich ist.
Therapieplanung
Nachdem die Diagnose feststeht, erfolgt die Therapieplanung. Um eine möglichst individuelle, auf den Patienten zugeschnittene (risikoadaptierte) Behandlung durchführen zu können, berücksichtigt das Behandlungsteam bei der Planung bestimmte Faktoren, die die Prognose des Patienten beeinflussen (so genannte Risiko- oder Prognosefaktoren).
Besonders wichtig in diesem Zusammenhang ist das Krankheitsstadium des Patienten. Es wird daran bemessen, wie weit sich der Tumor zum Zeitpunkt der Diagnose im Körper ausgebreitet hat und wie gut er bei einer Operation entfernt werden kann (siehe Tabelle zur Stadieneinteilung im Anschluss). Weitere wichtige Prognosefaktoren sind das Alter des Patienten sowie die feingeweblichen und, vor allem, die molekulargenetischen Eigenschaften des Tumors, die Aufschluss über sein Wachstums- und Metastasierungsverhalten geben können (siehe Abschnitt "Diagnose"). All diese Faktoren fließen in die Behandlungsplanung ein mit dem Ziel, für jeden Patienten durch die Auswahl der jeweils adäquaten Therapie das bestmögliche Behandlungsergebnis zu erreichen.
Mehr zu den Krankheitsstadien des Neuroblastoms finden Sie im Anschluss. Informationen zu weiteren bei der Therapieplanung berücksichtigten Prognosefaktoren finden Sie hier.
Krankheitsstadien
Die Ausbreitung des Tumors im Körper beeinflusst in der Regel deutlich die Heilungsaussichten und ist somit ein wichtiges Kriterium bei der Wahl der geeigneten Behandlungsstrategie. Die Einteilung des Neuroblastoms nach Krankheitsstadien berücksichtigt zunächst die Größe des Tumors, die Beteiligung von Lymphknoten sowie das Vorhandensein von Metastasen. Weitere Kriterien, die bei der Stadieneinteilung (Klassifikation) eine Rolle spielen, hängen von der verwendeten Klassifikation ab. Zwei Klassifikationssysteme werden derzeit parallel verwendet:
- INSS-Klassifikation: Nach der traditionellen, in Deutschland lange Zeit üblichen internationalen Stadieneinteilung (International Neuroblastoma Staging System, INSS) wurde – zusätzlich zu den oben genannten Faktoren – auch das Ausmaß der Operation mit einbezogen; die exakte Beurteilung des Krankheitsstadiums war daher erst nach dem operativen Eingriff möglich.
- INRG-Klassifikation: Die oben erwähnte INSS-Klassifikation wird zwar noch berücksichtigt, international gültig ist jedoch inzwischen die Internationale Neuroblastom-Risikoklassifizierung (englisch: International Neuroblastoma Risk Group Staging System, INRG). Die INRG-Stadieneinteilung schätzt das Krankheitsstadium bereits vor der Operation anhand festgelegter Risikofaktoren ein, die mit bildgebenden Verfahren (MRT oder CT) sichtbar werden. Ein solcher Risikofaktor (englisch: Image Defined Risk Factor, kurz IDRF) ist zum Beispiel die Ummauerung großer Gefäße durch den Tumor. Neben der voraussichtlichen Operabilität des Tumors, dem Alter des Patienten und molekularen Tumoreigenschaften fließen auch die feingewebliche Zuordnung des Neuroblastoms in die Stadieneinteilung mit ein.
Im Anschluss stellen wir die beiden Stadieneinteilungen parallel vor. Die INSS-Klassifikation unterscheidet die lokal begrenzten Stadien 1-3, das metastasierte Stadium 4 und das metastasierte Säuglingsneuroblastom 4S. Die INRG-Stadieneinteilung unterscheidet die lokal begrenzten Stadien L1 und L2 unter Berücksichtigung bestimmter Risikofaktoren sowie die fortgeschrittenen (metastasierten) Stadien M und MS (siehe Tabelle im Anschluss).
|
INSS Stadien |
Definition |
INRG Stadien |
Definition |
|---|---|---|---|
|
1 |
Vollständig entfernter Tumor |
L1 |
Lokalisierter Tumor ohne Nachweis von IDRF und begrenzt auf eine Körperhöhle |
|
2a |
Nicht vollständig entfernter Tumor Befall nur auf einer Seite der Wirbelsäule kein Lymphknotenbefall in der Umgebung des Tumors |
||
|
2b |
Vollständig oder unvollständig entfernter Tumor Befall nur auf einer Seite der Wirbelsäule benachbarte Lymphknoten auf der gleichen Körperseite sind befallen |
L2 |
Lokalisierter Tumor mit Nachweis von einem oder mehreren IDRF |
|
3 |
Nicht vollständig entfernter Tumor mit Wirbelsäulenüberschreitung oder Befall von Lymphknoten auf der dem Tumor gegenüberliegenden Körperseite |
||
|
4 |
Vorliegen von Fernmetastasen (zum Beispiel in Knochenmark, Knochen, Leber, Haut, entfernten Lymphknoten und anderen Organen) |
M |
Nachweis von Fernmetastasen (außer Stadium MS) |
|
4S |
nur bei Säuglingen unter 1 Jahr bei Diagnosestellung: Lokaler Tumor (Stadium 1, 2A oder 2B) mit Metastasen begrenzt auf Haut, Leber und/oder, minimal, im Knochenmark |
MS |
Metastatische Erkrankung bei Kindern im Alter von unter 18 Monaten mit Metastasen begrenzt auf Haut, Leber und Knochenmark. |
Mit Ausnahme der Patienten mit Stadium 4S beziehungsweise MS haben Patienten mit weniger fortgeschrittener Erkrankung in der Regel eine bessere Prognose als Patienten in fortgeschrittenen Krankheitsstadien (dazu gehören zum Beispiel die Stadien 3 und 4). Patienten mit weniger günstigen Heilungsaussichten bedürfen in der Regel einer intensiveren Therapie als Patienten mit günstiger Prognose (siehe Abschnitt „Behandlungsablauf“).
Krankheitsverläufe
Der Krankheitsverlauf eines Neuroblastoms ist individuell verschieden, bedingt vor allem durch das Wachstumsverhalten des Tumors und das Maß seiner Ausbreitung zum Zeitpunkt der Diagnose. So kann ein Neuroblastom bei Diagnosestellung auf seinen Ursprungsort begrenzt sein, es kann aber auch bereits Gewebe und Lymphknoten in der Umgebung befallen oder sich in weiter entfernten Organen angesiedelt haben (siehe auch Abschnitt "Krankheitsstadien" oben). Eine Besonderheit der biologisch und klinisch günstigen Neuroblastome ist, dass sie spontan zu gutartigen Tumoren ausreifen (so genannte Differenzierung) oder sich spontan zurückbilden können (spontane Regression).
Tumorwachstum und Metastasierung
Vor allem bei Kindern jenseits der ersten 18 Lebensmonate wachsen Neuroblastome oft rasch und ungehemmt und verbreiten sich – meist über das Blutsystem, manchmal aber auch über das Lymphsystem [lymphatisches System] – im gesamten Körper. Es bilden sich Tochtergeschwülste (Metastasen), vorwiegend in Knochenmark (bei 90 % der Patienten), Knochen (60 %), entfernten Lymphknoten (20 %) und Leber (17 %), seltener in Gehirn (9 %), Haut (2 %) und Lunge (1 %). In diesen Fällen handelt es sich um das Krankheitsstadium 4 beziehungsweise M.
Tumorausreifung (Differenzierung)
Manche Neuroblastome können – entweder spontan oder infolge einer Chemotherapie – reife und somit weniger bösartige Tumorzellanteile entwickeln. Man bezeichnet diesen Prozess als Tumorausreifung oder Differenzierung. Die entsprechenden Tumoren werden "Ganglioneuroblastome" genannt. Sie enthalten zwar noch immer bösartige Zellen, wachsen aber entschieden langsamer als die rein bösartigen Neuroblastome. Eine solche spontane Tumorausreifung wird gelegentlich bei Patienten nach dem ersten Lebensjahr beobachtet. Vollständig ausgereifte Ganglioneurome findet man meist erst nach dem vierten Lebensjahr oder im Erwachsenenalter.
Tumorrückbildung (spontane Regression)
Es gibt eine hohe Anzahl von Neuroblastomen, die sich spontan zurückbilden (Tumorregression). Die Tumorzellen sterben dabei durch eine Art selbstausgelösten Zelltod ab, ein Vorgang, den die Wissenschaftler Apoptose nennen.
Die spontane Tumor-Rückbildung wird vor allem und nahezu regelhaft bei Neuroblastomen beobachtet, die im Säuglings- und frühen Kindesalter auftreten und dem Tumorstadium 4S (beziehungsweise MS) zugeordnet werden. Bei diesen Patienten führt oft eine Lebervergrößerung infolge ausgedehnter Metastasen zur Diagnose. Diese Metastasen können zunächst noch rasch an Größe zunehmen, dabei Bauchorgane und Lunge verdrängen und ein lebensbedrohliches Ausmaß erreichen. Dann können sie sich aber spontan oder nach einer mild dosierten Chemotherapie zurückbilden. Spontane Tumor-Regressionen kommen allerdings nicht nur im Tumorstadium 4S (MS) bei Säuglingen vor, sondern können auch bei älteren Kindern mit lokalen Neuroblastomen der Stadien 1 bis 3 (beziehungsweise L1 bis L2) beobachtet werden.
Informationen zu den verschiedenen Krankheitsstadien eines Neuroblastoms erhalten Sie im Abschnitt "Therapieplanung" (siehe oben).
Behandlung
Die Behandlung eines Patienten mit Neuroblastom muss in einer kinderonkologischen Behandlungseinrichtung erfolgen. Dort ist das hoch qualifizierte Fachpersonal (Ärzte, Fachpflegekräfte) auf die Behandlung krebskranker Kinder spezialisiert und mit den modernsten Therapieverfahren vertraut. Darüber hinaus stehen die Ärzte dieser Klinikabteilungen in fachorientierten Arbeitsgruppen in ständiger, enger Verbindung miteinander und behandeln ihre Patienten nach gemeinsam entwickelten und stetig weiter verbesserten Therapieplänen.
Das Ziel der Behandlung ist, eine Heilung des Patienten zu erreichen und dabei das Risiko therapiebegleitender Nebenwirkungen und Spätfolgen so gering wie möglich zu halten.
Behandlungsmethoden
Die Therapie eines Neuroblastom-Patienten richtet sich nach seiner individuellen Krankheitssituation und der Wahrscheinlichkeit eines Krankheitsrückfalles. Bei manchen Patienten kann eine alleinige Operation zur Entfernung des Tumors oder zur Gewinnung einer Gewebeprobe ausreichend sein, bei anderen müssen mehrere Therapiemethoden miteinander kombiniert werden, um die Heilungschancen zu verbessern.
Die im Folgenden dargestellten Therapieformen stellen den gegenwärtigen Standard der Behandlung in Deutschland dar. Im Rahmen von klinischen Studien werden laufend neuere Therapiekonzepte überprüft, so dass die Behandlung einzelner Patienten unter Umständen von diesem Standard abweichen kann.
Zu den bei einem Neuroblastom eingesetzten Behandlungsmethoden gehören die Operation, die Chemotherapie und die Strahlentherapie. Bei Patienten mit einem besonders hohen Rückfallrisiko werden darüber hinaus eine Hochdosis-Chemotherapie mit nachfolgender autologer Stammzelltransplantation sowie eine Immuntherapie mit Antikörpern durchgeführt. Weitere Therapieverfahren können bei diesen Patienten ergänzend zum Einsatz kommen, zum Beispiel die MIBG-Therapie, das ist eine Behandlung mit radioaktiv markiertem Methyljodbenzylguanidin.
Die Operation zielt auf die Entfernung des Tumors und/oder die Gewinnung einer Gewebeprobe. Bei der Chemotherapie werden Medikamente verabreicht, die das Zellwachstum hemmen (Zytostatika) und so zur Vernichtung des Tumors beitragen. In der Regel werden Kombinationen verschiedenartig wirkender Zytostatika eingesetzt (Polychemotherapie), um die größtmögliche Wirkung gegen die bösartigen Zellen zu erzielen. Noch intensiver ist die Hochdosis-Chemotherapie: Sie zerstört nicht nur die Krebszellen, sondern auch das blutbildende System im Knochenmark, so dass als Ersatz anschließend patienteneigene Blutstammzellen transplantiert werden müssen. Eine bei Hochrisiko-Patienten erforderliche Strahlentherapie erfolgt mit energiereichen, elektromagnetischen Strahlen, die von außen durch die Haut auf die betroffene Region eingestrahlt werden (perkutane Strahlentherapie). Sie verursachen Schäden im Erbgut der Tumorzellen und führen dadurch zu deren Absterben.
Welche der genannten Verfahren in Frage kommen und in welcher Kombination, hängt in erster Linie von der Ausdehnung und Operabilität des Tumors, seinem Wachstumsverhalten sowie vom Alter des Patienten ab. Je weiter die Krankheit fortgeschritten ist und je größer das Risiko eines aggressiven Tumorwachstums oder auch eines Krankheitsrückfalles nach erfolgter Therapie ist, umso komplexer und intensiver wird letztlich die Therapie sein.
Da die Behandlung eines Neuroblastoms mit Nebenwirkungen einhergehen kann, erfolgen während der Behandlung unterstützende Therapiemaßnahmen (Supportivtherapie), die der Vorbeugung und/oder Behandlung dieser Begleiteffekte dienen. Hier finden Sie Informationen zur Supportivtherapie sowie Empfehlungen für zu Hause. Ausführliche Informationen zu den verschiedenen Therapieverfahren erhalten Sie hier.
Behandlungsabläufe
Jeder Patient wird zu Beginn der Behandlung einer bestimmten Risiko- oder Therapiegruppe zugeordnet. Die aktuellen Behandlungsrichtlinien sehen eine Einteilung in drei Therapiegruppen vor: Niedrige Risikogruppe, Mittlere Risikogruppe und Hochrisikogruppe. Für jede dieser Therapiegruppen gelten unterschiedliche Therapiepläne. Auf diese Weise kann eine auf den einzelnen Patienten abgestimmte, risikoangepasste Behandlung erfolgen.
Behandlung in der niedrigen Risikogruppe (Beobachtungspatienten)
In der niedrigen Risikogruppe (Beobachtungsgruppe) werden Patienten behandelt, die aufgrund eines lokalisierten Tumorwachstums und/oder ihres Alters durch die abwartende Haltung dieser Therapiestrategie nicht gefährdet werden. Ganz entscheidend für die Zuordnung eines Patienten zur Beobachtungsgruppe ist, dass keine ungünstigen molekulargenetischen Tumoreigenschaften (wie die MYCN-Amplifikation oder teilweise auch die 1p-Deletion) vorliegen. Zur Beobachtungsgruppe gehören demnach Patienten mit einer der folgenden Krankheitssituationen:
- Krankheitsstadium 1 (INSS), Alter 0-21 Jahre, keine MYCN-Amplifikation
- Krankheitsstadium 2 (INSS), Alter 0-21 Jahre, weder MYCN-Amplifikation noch 1p-Deletion
- Krankheitsstadium 3 (INSS), Alter 0-2 Jahre, weder MYCN-Amplifikation noch 1p-Deletion
- Krankheitsstadium 4S (INSS), Altersbegrenzung jedoch gemäß INRG-Stadium MS ausgedehnt auf 0-18 Monate, weder MYCN-Amplifikation noch 1p-Deletion
Behandlungsablauf: Bei Patienten mit einem Niedrigrisiko-Neuroblastom beschränkt sich die Behandlung in der Regel auf eine Operation zur Entfernung des Tumors; häufig ist aufgrund der hohen Rate an spontanen Tumorrückbildungen auch nur eine Gewebeentnahme (Biopsie) erforderlich. Bei gutem Allgemeinzustand der Patienten erfolgen keine Chemotherapie und keine Strahlentherapie. Allerdings wird der Krankheitsverlauf der Patienten im Rahmen engmaschiger Kontrolluntersuchungen (mittels regelmäßiger klinischer Untersuchung, Ultraschall, Magnetresonanztomographie, Tumormarker) weiter beobachtet. Im ersten Jahr werden die Patienten mindestens alle sechs Wochen, im zweiten bis fünften Jahr mindestens alle drei Monate und danach mindestens alle sechs bis zwölf Monate untersucht. Die Art der Untersuchungen hängt davon ab, ob die Patienten einen Resttumor haben und ob der Tumor mit Ultraschall gut darstellbar ist.
Wenn der Resttumor während der ersten zwölf Monate nach der Operation beziehungsweise bis zum zweiten Geburtstag erneut wächst und/oder mit Symptomen einhergeht, die einer Behandlung bedürfen (dazu zählen zum Beispiel ein schlechter Allgemeinzustand des Patienten, Ernährungsprobleme, Gewichtsverlust, Bluthochdruck, Harntransportstörungen), wird in der Regel eine milde Chemotherapie verabreicht, um eine Rückbildung des Tumors auszulösen. Dies gilt auch dann, wenn nach einer Tumorentfernung die Krankheit erneut auftritt (Krankheitsrückfall). Die Behandlung besteht aus bis zu vier Zyklen der Zytostatika-Kombination Doxorubicin, Vincristin und Cyclophosphamid. Alternativ können auch Carboplatin und Etoposid zum Einsatz kommen. Die Chemotherapie wird beendet, sobald das Tumorwachstum gestoppt ist. Bei manchen Patienten kann auch ein (weiterer) chirurgischer Eingriff zwecks Tumorentfernung oder zur Symptomentlastung angezeigt sein. Letzteres gilt zum Beispiel für Patienten mit dem Krankheitsstadium 4S, deren Tumor vor der Rückbildung zunächst noch stark wachsen kann.
Behandlung in der mittleren Risikogruppe
In der mittleren Risikogruppe (intermediäres Risiko) werden Patienten mit weiter fortgeschrittener Erkrankung und/oder höherem Lebensalter sowie bestimmten ungünstigen molekulargenetischen Eigenschaften (1p-Deletion) behandelt. Eine MYCN-Amplifikation muss ausgeschlossen sein. Zur mittleren Risikogruppe zählen somit Patienten mit einer der folgenden Krankheitssituationen:
- Krankheitsstadium 2 (INSS), Alter 0-21 Jahre, mit Veränderung in Chromosom 1p, aber ohne MYCN-Amplifikation
- Krankheitsstadium 3 (INSS), Alter 0-21 Jahre, mit Veränderung in Chromosom 1p, keine MYCN-Amplifikation
- Krankheitsstadium 3 (INSS), Alter 2-21 Jahre, weder MYCN-Amplifikation noch 1p-Deletion
- Krankheitsstadium 4 (INSS), Altersbegrenzung jedoch gemäß INRG-Stadium MS ausgedehnt auf 18 Monate, ohne MYCN-Amplifikation
Behandlungsablauf: Die Behandlung besteht aus der Operation oder, wenn das nicht möglich ist, zunächst einer Biopsie). Im Anschluss wird eine Chemotherapie durchgeführt. Sie setzt sich aus sechs Blöcken intensiver Chemotherapie (Induktionschemotherapie) und vier Blöcken einer etwas milderen Erhaltungschemotherapie zusammen. Sofern vor Beginn der Chemotherapie nur eine Biopsie entnommen werden konnte, wird nach den ersten Zyklen der Induktionschemotherapie eine operative Entfernung des Tumors angestrebt, denn meist verkleinert sich das Neuroblastom während der Chemotherapie.
Im Rahmen der Induktionstherapie werden standardmäßig abwechselnd Zytostatikakombinationen aus Doxorubicin, Vincristin und Cyclophosphamid beziehungsweise Carboplatin, Etoposid und Vindesin eingesetzt. Die Medikamentengabe erfolgt in Form mehrstündiger oder mehrtägiger Infusionen. Die Erhaltungstherapie besteht aus Cyclophosphamid, das meist in Tablettenform verabreicht wird.
Wenn nach der intensiven Chemotherapie noch ein aktiver Tumorrest zu finden ist, erfolgt – bei Kindern über 18 Monaten – parallel zur Erhaltungs-Chemotherapie eine Bestrahlung dieses Resttumors (mit einer Strahlendosis von 36 bis 40 Gy). Möglicherweise kann sich durch die Chemotherapie ein zuvor verbliebener Resttumor (zum Beispiel, wenn anfangs nur eine Biopsie erfolgte) auch so sehr verkleinern, dass er während oder im Anschluss an diese Therapiephase durch einen weiteren chirurgischen Eingriff vollständig entfernt werden kann. Die Gesamtdauer der Behandlung beläuft sich auf etwa ein Jahr.
Behandlung in der Hochrisiko-Gruppe
In der Hochrisiko-Gruppe werden alle Patienten im Krankheitsstadium 1, 2, 3 oder 4S behandelt, deren Tumor eine MYCN-Amplifikation aufweist, sowie alle Patienten mit Krankheitsstadium 4 ab einem Alter von 18 Monaten. Das Behandlungskonzept für Patienten mit Hochrisiko-Neuroblastom ist sehr umfangreich.
Behandlungsablauf: Nach der Operation oder Biopsie wird zunächst eine intensive, etwa fünfmonatige Chemotherapie mit mehreren Substanzen durchgeführt (so genannte Induktionschemotherapie). Die derzeitige Standard-Induktionstherapie beinhaltet sechs Chemotherapieblöcke unter Einsatz alternierender Zytostatikakombinationen aus Cisplatin, Etoposid und Vindesin beziehungsweise Vincristin, Dacarbazin, Ifosfamid und Doxorubicin. Zwischen oder nach den Chemotherapiezyklen erfolgt in der Regel die (Zweit-)Operation mit möglichst kompletter Entfernung des Tumors. Anschließend erhalten alle Patienten eine Hochdosis-Chemotherapie, auf die eine autologe Stammzelltransplantation folgt (Dauer: circa sechs Wochen). Bei Patienten mit MIBG-positivem Resttumor kann in Kombination mit der Hochdosistherapie zusätzlich eine Behandlung mit radioaktiv markiertem Methyljodbenzylguanidin (131-I-MIBG-Therapie) erfolgen. Die I-MIBG-Therapie findet in diesem Fall noch vor der Hochdosis-Chemotherapie statt.
Im Anschluss an die Hochdosisbehandlung folgen eine Bestrahlung des Tumorbetts und eine Immuntherapie mit dem Antikörper Dinutuximab beta. Ziel dieser Therapiephase (auch Erhaltungs- oder Post-Konsolidierungstherapie genannt) ist, eventuell verbliebene Tumorzellen zu vernichten. Wenn ein aktiver Resttumor vorliegt, wird eine Strahlendosis von bis zu 36 Gy empfohlen. Die Gesamt-Therapiedauer kann bis zu zwei Jahren dauern.
Anmerkung zur Studie HR-NBL2 für Patienten mit Hochrisiko-Neuroblastom: Im Rahmen der seit 2023 eröffneten Studie für Hochrisiko-Patienten werden sowohl für die Induktionschemotherapie als auch die Hochdosis-Chemotherapie und Strahlentherapie jeweils zwei unterschiedliche Therapiekonzepte in je zwei unterschiedlichen Behandlungsarmen miteinander verglichen. Die Zuordnung der Patienten zu dem einen oder anderen Therapiearm erfolgt zufällig (randomisiert) [siehe Randomisierung], sofern die jeweilige Behandlung in Frage kommt.
Therapieoptimierungsstudien und Register
Fast alle Kinder und Jugendliche mit einem Neuroblastom werden in Deutschland im Rahmen von Therapieoptimierungsstudien oder Registern behandelt. Therapieoptimierungsstudien sind kontrollierte klinische Studien, die darauf abzielen, erkrankte Patienten nach dem jeweils aktuellsten Wissensstand zu behandeln und gleichzeitig die Behandlungsmöglichkeiten zu verbessern und weiterzuentwickeln.
Patienten, die an keiner Studie teilnehmen, entweder, weil zum Zeitpunkt ihrer Erkrankung keine Studie verfügbar ist oder weil sie die Einschlusskriterien einer bestehenden Studie nicht erfüllen, werden oft in einem so genannten Register dokumentiert. Diese dienen zunächst dazu, die Therapie der Patienten wissenschaftlich zu begleiten. Zur Sicherung der optimalen Behandlung verfasst darüber hinaus die jeweilige Studiengruppe in der Regel detaillierte Empfehlungen und berät die behandelnden Ärzte bei der Auswahl der optimalen Therapie für den einzelnen Patienten.
Derzeit stehen in Deutschland (mit internationaler Beteiligung) folgende Therapieoptimierungsstudien und Register für Patienten mit Neuroblastom zur Verfügung:
- Studie HR-NBL2 (Hochrisiko Neuroblastom-Studie 2.0): Internationale, multizentrische, randomisierte Therapiestudie (Phase-III-Studie) für Patienten mit Hochrisiko-Neuroblastom. Die Studie der SIOP Europe Neuroblastoma (SIOPEN) wurde in Deutschland 2023 für die Patientenaufnahme eröffnet; die nationale Studienzentrale befindet sich an der Klinik für Pädiatrie m.S. Onkologie/Hämatologie der Charité - Universitätsmedizin Berlin unter der Leitung von Prof. Dr. med. Angelika Eggert. Die Studie vergleicht unterschiedliche Therapiekonzepte der Induktions-Chemotherapie sowie der Hochdosis-Chemotherapie und Strahlentherapie mit dem Ziel, die Prognose der Patienten weiter zu verbessern. Zahlreiche Behandlungseinrichtungen in ganz Europa sowie auch außerhalb Europas sind an der Studie beteiligt.
- NB Register 2016: Register für Neugeborene, Säuglinge, Kinder, Jugendliche (sowie Erwachsene), die neu an einem neuroblastischen Tumor (Neuroblastom, Ganglioneuroblastom, Ganglioneurom) erkranken oder einen Krankheitsrückfall erleiden. Das Register wurde 2017 nach Beendigung der Therapiestudien NB 2004 und NB 2004-HR eröffnet und dient insbesondere dazu, Kenntnisse über Häufigkeit, Krankheitsverlauf und Langzeitfolgen der Erkrankung zu erlangen und die Prognose zu verbessern. Die Entscheidung, welche Therapie durchgeführt wird, trifft der behandelnde Arzt, unterstützt durch Therapieempfehlungen von Seiten der Studienzentrale. Die Meldung in das Register schließt eine (zukünftige) Beteiligung an einer klinischen Studie nicht aus. Die Registerzentrale befindet sich an der Universitäts-Kinderklinik in Köln unter der Leitung von Prof. Dr. med. Thorsten Simon.
Anmerkung zu Rezidivstudien: Für Patienten mit Rückfall (Rezidiv) oder Nichtansprechen eines Hochrisiko-Neuroblastoms stehen Phase-I/II-Studien zur Verfügung, über die die Studienleitungen in Berlin, Köln und Greifswald stets aktuell informieren können. Kontaktdaten zu den Studienzentralen Köln, Berlin und Greifswald finden Sie hier.
Prognose
Die Heilungsaussichten lassen sich bei einem Neuroblastom für den Einzelfall nur schwer abschätzen. Sowohl das Ausmaß der Erkrankung als auch die Aggressivität des Tumors und das Alter des Patienten spielen eine Rolle. Eine sehr gute Prognose – mit 10-Jahres-Überlebensraten von zum Teil weit über 90 % – besteht bei Kindern mit dem Neuroblastom-Stadium 4S sowie in der Regel bei Patienten mit begrenzten Tumoren (Stadium 1 und 2). Auch jüngere Kinder (unter 18 Monaten) mit Krankheitsstadium 3 haben, sofern keine ungünstigen molekularen Tumoreigenschaften vorliegen, eine gute Prognose. Bei älteren Kindern mit metastasiertem Neuroblastom (Stadium 4) sind die Heilungsaussichten trotz intensiver Therapie mit maximal 50 % noch immer ungünstig.
Anmerkung: Bei den genannten Überlebensraten handelt es sich um statistische Größen. Sie stellen nur für die Gesamtheit der an einem Neuroblastom erkrankten Patienten eine wichtige und zutreffende Aussage dar. Ob der einzelne Patient geheilt werden kann oder nicht, lässt sich aus der Statistik nicht vorhersagen. Wenn Sie Fragen zur prognostischen Einschätzung der Erkrankungsart Ihres Kindes haben, wenden Sie sich daher bitte an Ihr Behandlungsteam.
Weitere Informationen
Die hier vermittelten Informationen sind vor allem auf der Grundlage der unten angegebenen Literatur, unter Berücksichtigung der aktuellen Leitlinien und Therapiepläne zur Behandlung von Kindern und Jugendlichen mit Neuroblastom und in Zusammenarbeit mit der Neuroblastom-Studienzentrale erstellt worden. Weitere Informationen zum Thema erhalten Sie im ausführlichen Patiententext zum Neuroblastom in unserem Informationsportal www.kinderkrebsinfo.de. Bei weiteren Fragen können Sie jederzeit Ihren behandelnden Arzt ansprechen.
 PDF-Datei der Patienten-Kurzinformation zum Neuroblastom (356KB)
PDF-Datei der Patienten-Kurzinformation zum Neuroblastom (356KB)Literatur 
- Erdmann F, Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Annual Report 2019 (1980-2018). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2020 [URI: https://www.kinderkrebsregister.de/ typo3temp/ secure_downloads/ 42507/ 0/ 1c5976c2ab8af5b6b388149df7182582a4cd6a39/ Buch_DKKR_Jahresbericht_2019_komplett.pdf]
- Simon T: Leitlinie: Neuroblastom. S1-Leitlinie 025-008 (Leitlinie der Gesellschaft für Pädiatrische Onkologie und Hämatologie) AWMF-online 2019 [URI: https://www.awmf.org/ uploads/ tx_szleitlinien/ 025-008l_S1_Neuroblastom_2019-07_01.pdf]
- Eggert A, Simon T, Hero B, Lode H, Ladenstein R, Fischer M, Berthold F: Neuroblastom. in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie Springer Verlag GmbH GDeutschland 2006, 2018, 2. vollständig überarbeitete Auflage 2018, 420 [ISBN: 978-3-662-43685-1]
- Berthold F, Spix C, Kaatsch P, Lampert F: Incidence, Survival, and Treatment of Localized and Metastatic Neuroblastoma in Germany 1979-2015. Paediatric drugs 2017, 19: 577 [PMID: 28786082]
- Fischer J, Pohl A, Volland R, Hero B, Dübbers M, Cernaianu G, Berthold F, von Schweinitz D, Simon T: Complete surgical resection improves outcome in INRG high-risk patients with localized neuroblastoma older than 18Â months. BMC cancer 2017 Aug 4; 17: 520 [PMID: 28778185]
- Simon T, Hero B, Schulte JH, Deubzer H, Hundsdoerfer P, von Schweinitz D, Fuchs J, Schmidt M, Prasad V, Krug B, Timmermann B, Leuschner I, Fischer M, Langer T, Astrahantseff K, Berthold F, Lode H, Eggert A: 2017 GPOH Guidelines for Diagnosis and Treatment of Patients with Neuroblastic Tumors. Klinische Padiatrie 2017, 229: 147 [PMID: 28561228]
- Oberthuer A, Berthold F, Hero B, Till H: Neuroblastome, in: Solide Tumoren im Kindesalter. Fuchs J (Hrsg.) Schattauer GmbH: Stuttgart 2012, 77 [ISBN: 978-3-7945-2786-1]
- Brisse HJ, McCarville MB, Granata C, Krug KB, Wootton-Gorges SL, Kanegawa K, Giammarile F, Schmidt M, Shulkin BL, Matthay KK, Lewington VJ, Sarnacki S, Hero B, Kaneko M, London WB, Pearson AD, Cohn SL, Monclair T, International Neuroblastoma Risk Group Project: Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology 2011, 261: 243 [PMID: 21586679]
- Øra I, Eggert A: Progress in treatment and risk stratification of neuroblastoma: impact on future clinical and basic research. Seminars in cancer biology 2011, 21: 217 [PMID: 21798350]
- Hero B, Papenheim H, Schuster U: Neuroblastom – Informationen für Eltern. Fördergesellschaft Kinderkrebs-Neuroblastom-Forschung e.V., Baden 2011 [URI: http://www.neuroblastoma.de/ fileadmin/ PDF/ Neuroblastom.pdf]
- Maris JM: Recent advances in neuroblastoma. The New England journal of medicine 2010 Jun 10; 362: 2202 [PMID: 20558371]
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, von Schweinitz D, Simon T, Cohn SL, Pearson AD, INRG Task Force: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. Journal of clinical oncology 2009, 27: 298 [PMID: 19047290]
- Oberthuer A, Theissen J, Westermann F, Hero B, Fischer M: Molecular characterization and classification of neuroblastoma. Future oncology (London, England) 2009, 5: 625 [PMID: 19519203]
- Fischer M, Spitz R, Oberthür A, Westermann F, Berthold F: Risk estimation of neuroblastoma patients using molecular markers. Klinische Padiatrie 2008, 220: 137 [PMID: 18478485]
- Hero B, Simon T, Spitz R, Ernestus K, Gnekow AK, Scheel-Walter HG, Schwabe D, Schilling FH, Benz-Bohm G, Berthold F: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. Journal of clinical oncology 2008, 26: 1504 [PMID: 18349403]
- Ebell W: Hämatopoetische Stammzelltransplantation. in: Gadner H, Gaedicke G, Niemeyer CH, Ritter J:. Pädiatrische Hämatologie und Onkologie Springer-Verlag, 2006, 66 [ISBN: 3540037020]
- Claviez A, Lakomek M, Ritter J, Suttorp M, Kremens B, Dickerhoff R, Harms D, Berthold F, Hero B: Low occurrence of familial neuroblastomas and ganglioneuromas in five consecutive GPOH neuroblastoma treatment studies. European journal of cancer (Oxford, England : 1990) 2004, 40: 2760 [PMID: 15648116]
- Berthold F, Hero B, Kremens B, Handgretinger R, Henze G, Schilling FH, Schrappe M, Simon T, Spix C: Long-term results and risk profiles of patients in five consecutive trials (1979-1997) with stage 4 neuroblastoma over 1 year of age. Cancer letters 2003, 197(1-2): 11 [PMID: 12880954]
- Hero B, Berthold F: Neuroblastom. Monatschr Kinderheilkd 2002, 150: 775 [DOI: 10.1007/s00112-002-0493-0]